Anika Abbott and Kaveri Dole, Andover High School, Andover, Massachusetts, United States
Reviewed on 3 May 2025; Accepted on 9 June 2025; Published on 27 October 2025
With help from the 2025 BioTreks Production Team.
Congenital Central Hypoventilation Syndrome (CCHS) is a rare genetic disorder that mainly affects breathing. Those with this disorder take shallow breaths and can even stop breathing, particularly while sleeping. These difficulties cause the need for patients to be supported with an external ventilator. While some patients only require the use of a ventilator while asleep, in extreme cases, the support of a ventilator is also needed during the day.
Unfortunately, CCHS is caused by mutations in the PHOX2B gene that control many other genes, and it is frequently associated with autism spectrum disorders (ASD). CCHS is an extremely rare disease with only about 2,000 globally known cases, classifying it as a rare disease since it affects less than 0.05% of the population. Considering that today there are over 10,000 known rare diseases in the human population, there is a 4% chance of a newborn carrying a rare disease. This makes rare diseases a common problem overall. Patients and caregivers affected by rare diseases face health, psychological, and financial challenges due to delayed and inaccurate diagnoses.
Based on the inefficiency of diagnosing with current traditional methods, this perspectives article proposes a quantitative polymerase chain reaction (qPCR) chip utilizing TaqMan technology (primers and fluorescent probes) as an accurate and fast genetic screening tool for rare diseases. Using this technology, just one blood sample from a person can be used to screen for multiple rare diseases. A qPCR can be designed for any disease for which the causative gene and the mutations are known. Many additional reactions can be added to the independent wells of a qPCR chip. The advantage of using independent reactions is to avoid cross-reaction and inaccurate results often obtained by doing multiple reactions in the same well. In this perspectives article, we propose a prototype qPCR chip for the CCHS causative gene, PHOX2B. We also propose adding the two genes SHANK3 and MECP2, as they are associated with ASD, and CCHS patients have a higher chance of being diagnosed with ASD.
Keywords: Rare Disease, qPCR Chip, Genetic Diagnosis, Scalable, TaqMan
Authors are listed in alphabetical order. Vandana Dole and Lindsey L'Ecuyer mentored the group. Please direct all correspondence to lindsey.lecuyer@andoverma.us.

Motivations
Rare diseases range from benign to life-threatening. Not only are they difficult to diagnose, but this process is lengthy and expensive (Breining, 2017). Many patients remain undiagnosed for a prolonged period of time, meaning that they may even die before receiving an accurate diagnosis. The wide range of different rare diseases makes accurate diagnosis difficult, sending patients on a long journey referred to as a “diagnostic odyssey” (Breining, 2017). Using technologies such as quantitative polymerase chain reaction (qPCR) chips with TaqMan probes, we aim to screen for numerous rare diseases simultaneously to reduce both the cost and time that goes into diagnosis compared to traditional methods that are inefficient for this population.
The specific rare diseases that this perspectives article focuses on are CCHS, an extremely rare neurological disorder that causes difficulty in self-regulating functions of the body, such as breathing, especially while asleep. It also affects heart rate, blood pressure, temperature control, and more (CCHS Network, 2025). ASD, otherwise known as autism, are complex disorders where, amongst other symptoms, patients’ behavior often becomes repetitive. Many CCHS patients also suffer from ASD, making the healthcare extraordinarily challenging for the patients and caregivers.
CCHS is just one example of a rare disease with a long and difficult diagnosis process. The arduous journey of diagnosis begins after the patient starts to display symptoms, including difficulty with breathing. This follows multiple days in the hospital and a series of tests, often eliminating many metabolic conditions before a confirmed diagnosis is obtained.
Examples of some better-known rare diseases are cystic fibrosis, Tay-Sachs disease, sickle cell disease, Down syndrome, and muscular dystrophy. If the diagnosis of all these rare diseases could be done cheaply, the standard of care could be to test everyone early in life. This could be revolutionary in helping to find these diseases early in the patients’ lives, avoiding complications from delayed diagnosis.
PHOX2B and CCHS
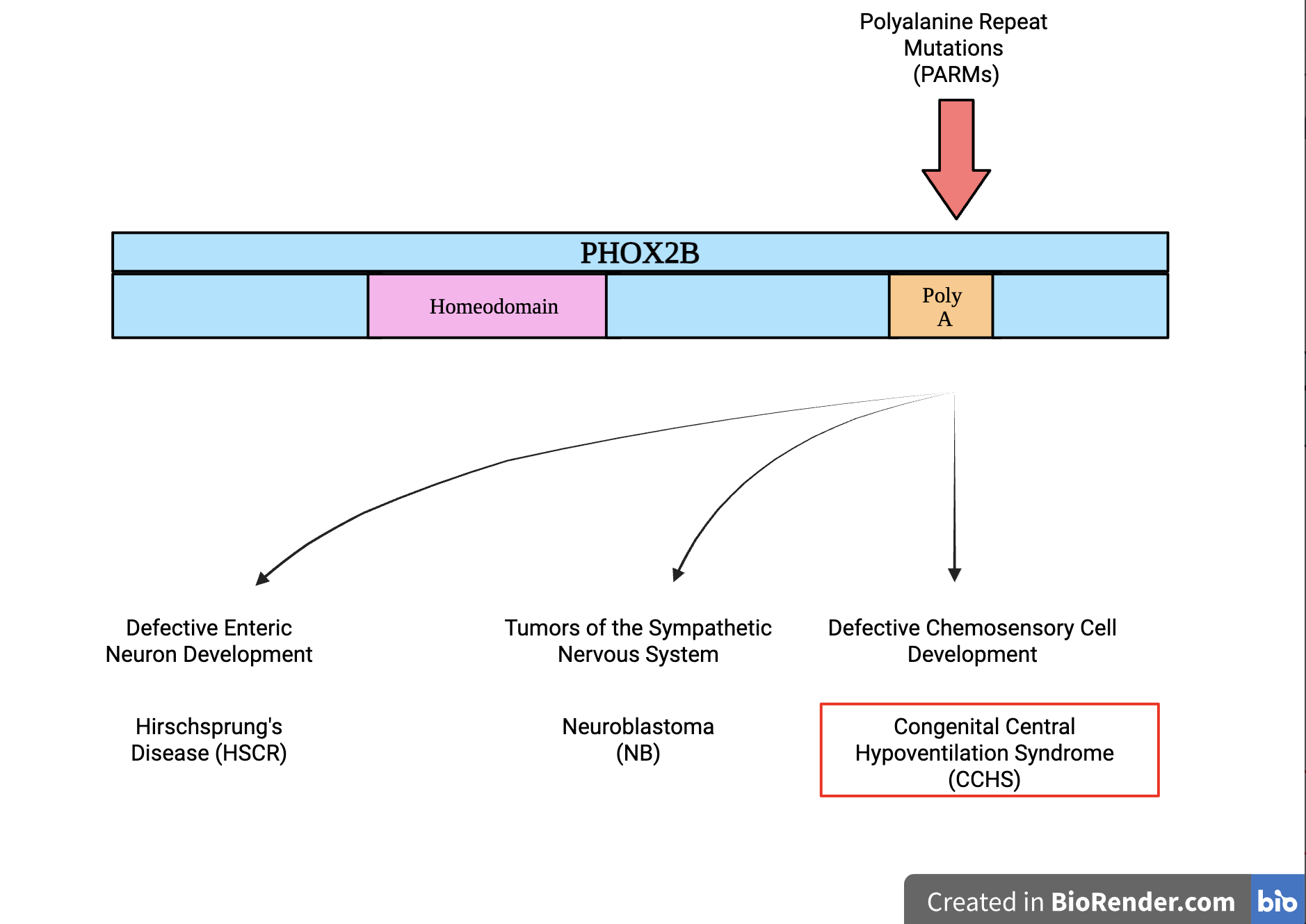
The wild-type Paired-like Homeobox 2B (PHOX2B) gene contains a sequence of 20 alanines in its code. However, due to spontaneous mutations in rare instances, the mutated PHOX2B genes contain more than 20 alanine stretches, causing CCHS.
The PHOX2B protein is a transcription factor in the paired homeobox family, meaning it has two DNA-binding areas: the paired domain and the homeodomain. Having two DNA-binding areas as opposed to one increases its transcription strength (Mark et al., 1997). PHOX2B is a transcription factor that binds to DNA and acts as a switch to turn other genes on and off to transcribe mRNA. These genes are mostly ones involved in the development of neurons, such as TH (tyrosine hydroxylase), DBH (dopamine beta-hydroxylase), RET, TLX-2, and numerous others. Since PHOX2B controls many genes, a mutation in PHOX2B causes multiple issues that are referred to as a syndrome, as shown in Figure 1. The most stark feature of CCHS is an inability to breathe when asleep, as the autonomic breathing function controlled by the brain is dysfunctional. Due to a mutation in PHOX2B, noradrenergic neurons are not formed properly. These neurons make and release norepinephrine, an adrenaline-like brain chemical vital for attention and alertness, stress responses, control of heart rate and blood pressure, breathing, and digestion. CCHS patients experience difficulties with all these functions (Cleveland Clinic, 2025).
| Figure 1. Different PHOX2B Mutations and How They Affect Patient Phenotypes. This figure shows the Polyalanine Repeat Mutations (PARMs) in the PHOX2B gene, which causes CCHS and other conditions, including Neuroblastoma and Hirschsprung’s Disease. In a wild-type PHOX2B gene’s code, there should be 20 polyalanine repeats; however, CCHS patients with PARMs have 35-44 polyalanines. (Chang, D. F. et al., 2021)). |
|
|---|
CCHS and ASD
CCHS and ASD are two different and distinct conditions; however, they are linked together through shared malfunctions in the Autonomic Nervous System (ANS), which is a part of the Peripheral Nervous System (PNS) that regulates involuntary processes such as heart rate and digestion (Waxenbaum, 2023). Particularly, both CCHS and ASD affect the Parasympathetic Nervous System (PSNS), which is a group of nerves in the PNS that regulates the activity of body functions such as breathing during periods of rest, like sleep (Cleveland Clinic, 2022). It was reported in one study from a French National Center that 8.7% of CCHS patients under 20 years old were also diagnosed with ASD (Dudoignon et al., 2024). CCHS is diagnosed by DNA sequencing to confirm the mutation in the PHOX2B gene, which is costly and inefficient. DNA sequencing is an expensive process, ranging from $100 to $2,000, and is not always accessible due to its cost, especially considering that it is often not covered by insurance. Additionally, from when a sample is received, it may take anywhere from a few days to several weeks to get a result (MedlinePlus, 2025). In contrast, genetic testing using qPCR is more cost effective, timely, and can test for multiple rare diseases simultaneously, as proposed here.
SHANK3, MECP2 and ASD
Two of the mutations associated with ASD are those found in the SH3 and multiple ankyrin repeat domains 3 (SHANK3) and Methyl-CpG binding protein 2 (MECP2) genes.
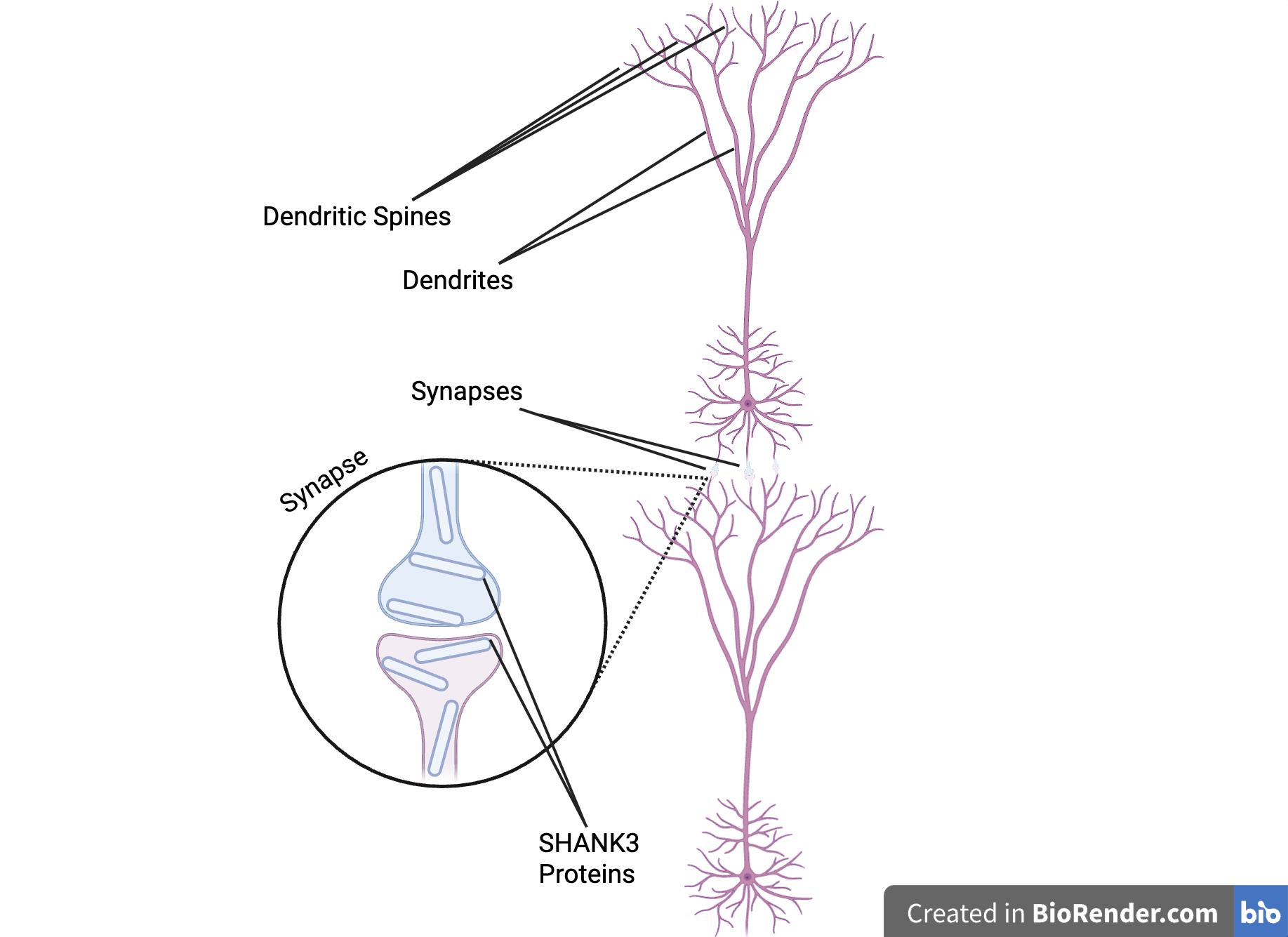
The SHANK3 protein, coded for by the SHANK3 gene, is a member of the SHANK protein group (National Library of Medicine, 2025a). This group is responsible for building and organizing synapses, which are small pockets of space between two neurons where the two neurons may conduct synaptic transmission, or communication. A single neuron may contain thousands of synapses (Dana Foundation, 2023), which is why SHANK3 proteins have such a big role in neurological function. Neurotransmitters are transmitted from one neuron to the other through the synapse. This happens when the presynaptic cell releases neurotransmitters from the synaptic vesicles near the cell membranes into the space between the two cells. These neurotransmitters are taken up by membrane receptors on the postsynaptic cell.Specifically, the SHANK3 proteins act as a support structure inside the synapse. They hold together neurotransmitter receptors to receive signals between synapses, ion channels controlling signal flow, and other proteins, as shown in Figure 2. They also link the previously stated parts to a cell’s skeleton and the signaling systems inside the cell. SHANK3 also supports the growth and structure of dendritic spines. Dendrites are projections from the neurons that receive input from presynaptic neurons or the environment. Dendritic spines are small bulbous protrusions on these dendrites that assist synaptic transmission (Hering and Sheng, 2001).
| Figure 2. The Role of SHANK3 Proteins in Neurons. This image shows two neurons, with their dendritic spines branching from their dendrites that are connected by a synapse with SHANK3 protein inside, providing stability to the synaptic structure (MIT News, 2018). |
|
|---|
A mutation in the SHANK3 gene at the c.2260C>T position results in a premature stop codon, due to which the SHANK3 protein is not formed (National Library of Medicine, 2025b). This mutation is associated with ASD (Sarowar, 2016).
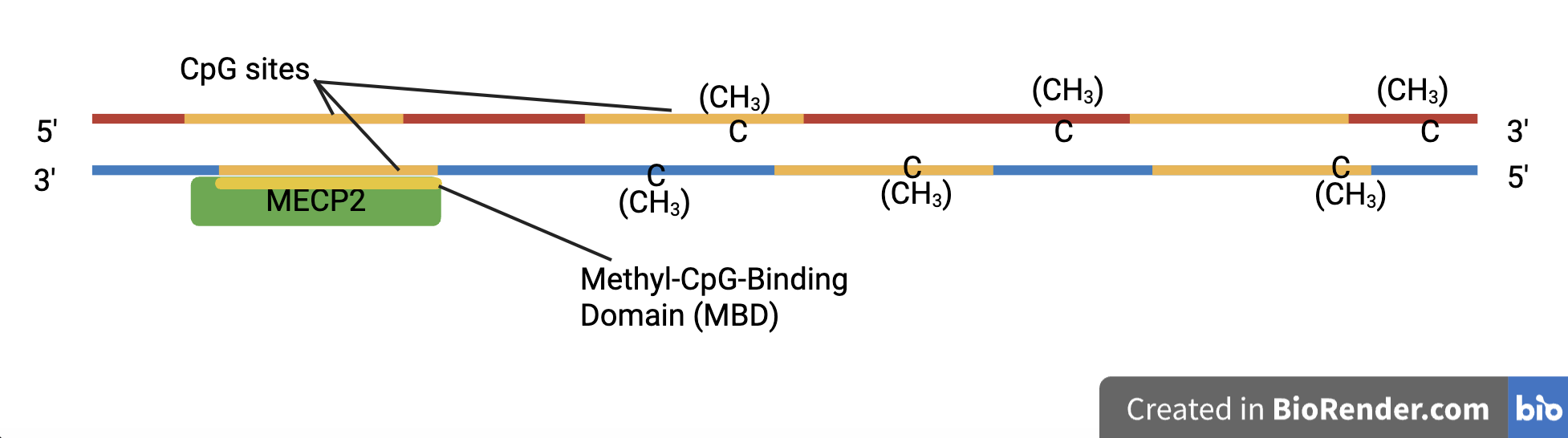
The MECP2 protein, coded for by the MECP2 gene, is an essential part of embryonic development. It specializes in identifying DNA methylation sites (Du et al., 2015).
| Figure 3. The Function of Methyl-CpG-Binding Domain (MBD) Proteins. This image shows the MECP2 protein, a member of the MBD protein family. It includes its MBD, which binds to the CpG sites on a methylated DNA strand. |
|
|---|
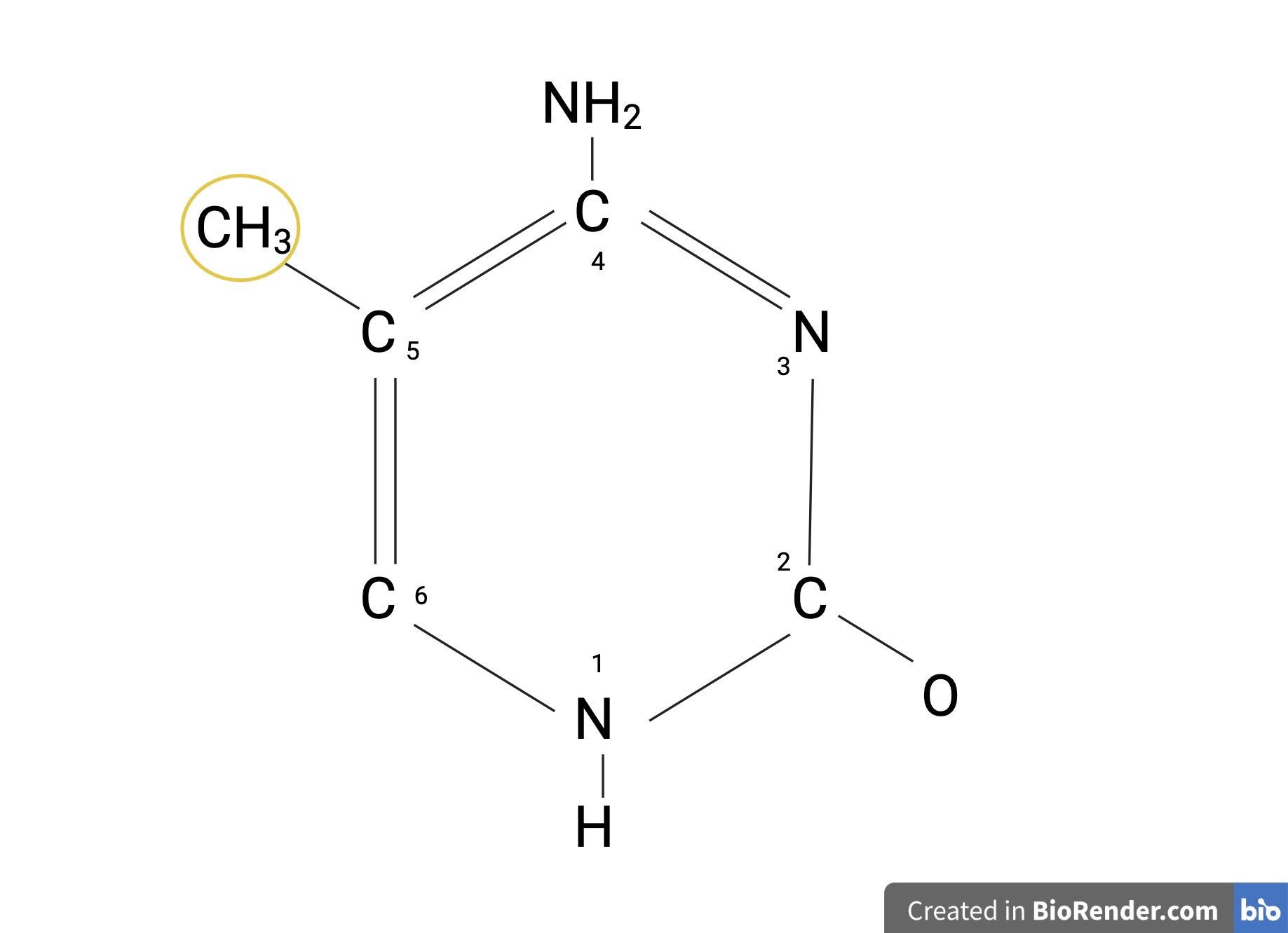
DNA methylation is an epigenetic mechanism that involves transferring or binding a methyl group, the compound CH3, onto the C5 position on the cytosine base to form 5-methylcytosine, as shown in Figure 4. DNA methylation regulates gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factors to DNA. (Moore et al., 2012).
| Figure 4. The Structure of 5-Methylcytosine. This is an image of 5-Methylcytosine with a methyl group (CH3) bound onto the C5 position of the cytosine structure. |
|
|---|
The 5-methylcytosine promotes or inhibits transcription factors from binding to the DNA sequence, altering gene activity (Moore et al., 2012). This influences the amount of RNA and proteins synthesized, as well as specific genes that can be turned on or off (Breiling 2015). Proteins in the MBD protein family, such as MECP2, have a sequence of about 70 amino acids in their code that selectively bind to methylated CpG sites (National Library of Medicine, 2025c). They use chromatin modeling complexes on methylated DNA, which may compact the chromatin and silence transcription.
A mutation in the MECP2 protein, while primarily linked with Rett syndrome, is also found in patients diagnosed with ASD. Either decreased or increased MECP2 expression or function, for example, MECP2 duplication syndrome, is found to lead to ASD (Nagarajan et al., 2007; Wen et al., 2017).
TaqMan
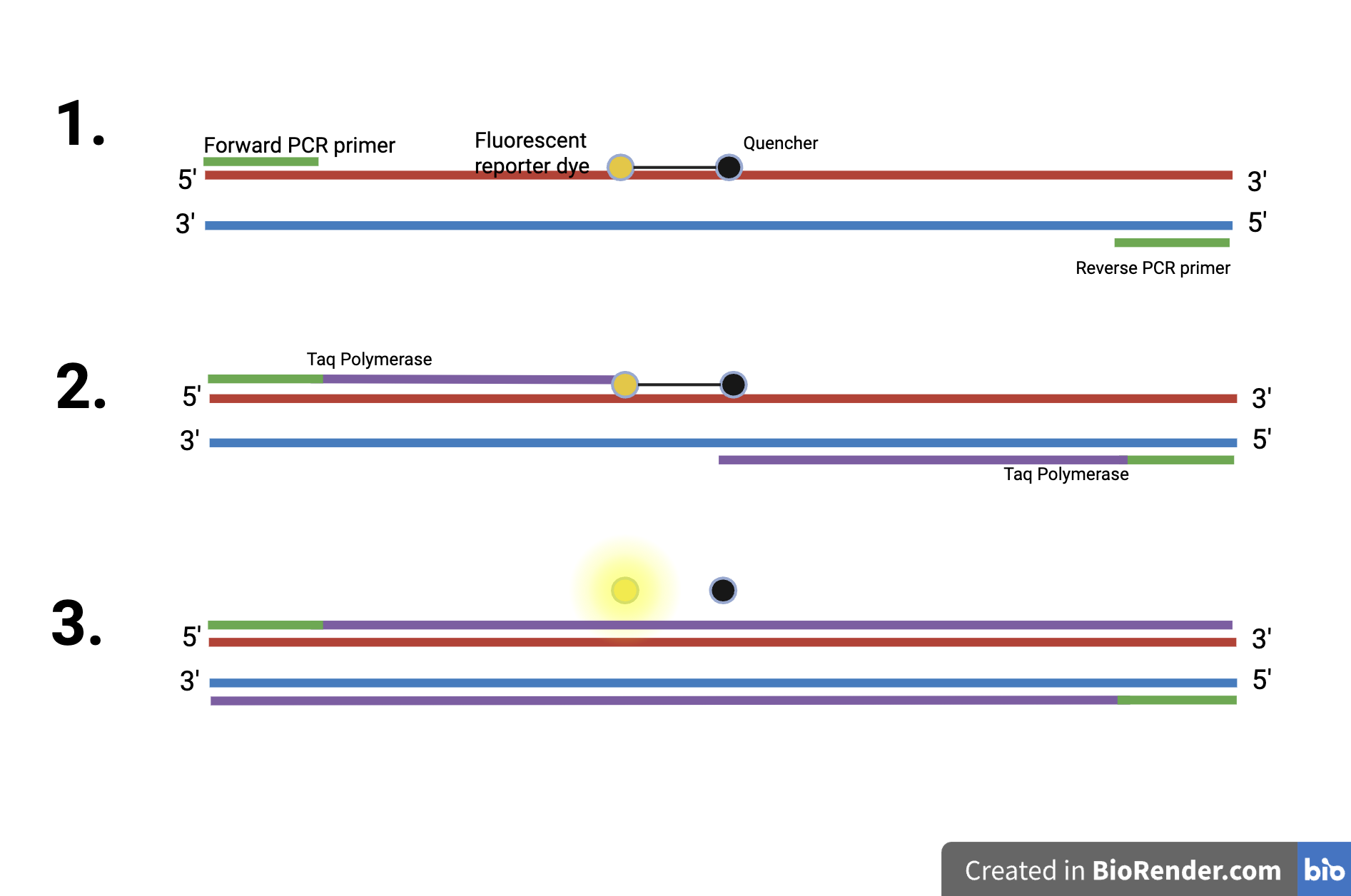
TaqMan refers to the technology used for detecting and quantifying DNA sequences using real-time quantitative PCR, or qPCR, as shown in Figure 5. It works by utilizing a probe, which is a short single-stranded DNA sequence that binds specifically to a target DNA. This probe has a fluorescent reporter dye on one end and a quencher on the other. The quencher inhibits the fluorescence from the reporter dye. The quencher prevents the reporter dye from emitting fluorescence when a target sequence is not present. This probe binds to the target DNA between the primers of both DNA strands. Then, the enzyme Taq polymerase starts DNA synthesis from the primers, adding nucleotides according to the PCR process. When the Taq polymerase reaches the probe, its 5’ to 3’ exonuclease activity cleaves the DNA probe in the middle. This separates the fluorescent reporter dye and the quencher, allowing the fluorescence to be detected since it is now free from the quencher. As more DNA is amplified, the fluorescence increases. This allows the qPCR machine to monitor the fluorescence and quantify the DNA in real time. Compared to PCR, having a probe in addition to the primers makes the TaqMan qPCR reaction highly specific.
| Figure 5. The TaqMan Process. This image shows the TaqMan technology process, where the 5′-3′ exonuclease activity of the Taq polymerase enzyme cleaves the probe’s fluorescent reporter dye and the quencher. This allows for fluorescence to be detected. Also, the PCR process is happening, where DNA is amplified. |
|
|---|
qPCR Chip
qPCR is a laboratory technique used to amplify and quantify DNA. It is similar to Polymerase Chain Reaction (PCR). It uses the same basic process of heating and cooling cycles to replicate a specific DNA segment many times. This cycle includes denaturation, where DNA is heated to separate its two strands. Then, annealing occurs, where the mixture is cooled in order for the primers to bind to the target region of the DNA. Lastly, Taq polymerase adds new DNA nucleotides to the primers to build the two new strands. This cycle is repeated, where the number of DNA strands is doubled each round. This allows for millions of copies of a specific DNA segment to be made accurately and within a few hours. With TaqMan qPCR, however, the amount of DNA is measured after each cycle using a fluorescent dye in a probe that binds to the DNA. When more DNA is produced, more fluorescence will be detected, allowing for the amount of DNA replicated to be quantified. Also, unlike PCR, qPCR results are detected in real-time, cycle by cycle, as DNA amplification happens.
In order to design a genetic diagnosis, the correlation between the mutation and disease phenotype must be well established. This study proposes the use of a qPCR chip to amplify and quantify targeted sequences of DNA that represent either the wild-type or the mutated gene associated with various rare diseases. It uses both primers and probes specific to the wild-type or mutated gene of a rare disease to distinguish the wild-type or mutant genes. The qPCR chip can detect the presence of these mutations in a sample of DNA and, therefore, can be used for the screening of rare diseases. The Thermo Fisher OpenArray qPCR chip is the size of a microscope slide and has a total of 3,072 wells. The approximate volume of a well of the qPCR chip is 33 nanoliters. The reaction wells are approximately 300 micrometers in length and width. As little as 0.6 mL of a blood sample can yield 30 micrograms of DNA, which is enough for 3,072 reactions on a chip. The individual wells of the qPCR chip contains primers and probes for either the wild type or the mutated gene. A panel of multiple rare diseases (wild type and mutations) can fit on a single chip, and allows for accurate and fast diagnoses. Once the chip has been loaded, the DNA sample flows through the microfluidic system and reaches the reaction wells of the chip. Once the prototype is built, additional wells with individual reactions can be added to diagnose additional rare diseases. As we use the chip to diagnose more and more diseases, it will become more cost-effective. Due to this scalable and cost-effective solution for rare disease diagnosis, eventually, we would be able to screen for more rare diseases concurrently. We begin by designing primers and probes to diagnose two diseases. We target mutations in the PHOX2B gene, which is associated with CCHS, and the SHANK3 and MECP2 genes associated with ASD, as shown in Figure 6.
| Figure 6. The Proposed qPCR Chip. A representation of the qPCR chip is shown here. Buffer (top) and blood sample (bottom) are loaded from their respective channels. Through microfluidic channels, the buffer and blood sample enter each independent well on the top and bottom panels. The buffer contains nucleotides, DNA polymerase, and other constituents needed for the qPCR reaction, except for the primers and probes. The blood sample contains DNA from the blood of the patient, along with the buffer. The buffer contains Taq polymerase, salts, and nucleotides required for the qPCR reaction, with the exception of primers and probes, which are pre-loaded into the wells. The wells of the top buffer panel are either negative controls or positive wild-type or positive mutant controls for the three genes, PHOX2B, SHANK3, and MECP2. In the top panel, when the buffer enters the wells, it starts the qPCR reaction when the wells are preloaded with primers, probes, and control DNA. No signals are expected to be detected from the negative control well with only primers and probes. The wells of the bottom blood sample panel are the wild-type primer and probe or the mutant primer and probe for the three genes. Upon the buffer and blood sample entering the wells, if the pre-loaded primers and probes match the DNA sequence, the qPCR reaction will take place, and a fluorescent signal will be detected. |

|
|---|
Development of positive controls
Positive controls used in our qPCR chip glow fluorescently in the qPCR reaction when the preloaded primers and probes match. The qPCR machine displays the result in real time. As the cycles are completed, the graph displayed shows the number of PCR cycles on the X-axis and the glow signal from the TaqMan probe on the Y-axis. The signal keeps increasing with the cycles until it plateaus when the TaqMan probe or other reaction components are completely consumed. It is generated as follows.
Positive control DNA sequences are developed by cloning the positive control DNA in a plasmid such as pUC57. Plasmids are circular DNA sequences that can be transformed or introduced by bacteria. Each bacterium contains 20-100 copies of the plasmid in one bacterial cell. Since bacterial cells can be easily grown in Luria Broth (LB), which is a nutrient medium in the laboratory, the DNA sequence can be amplified and preserved using cloning in plasmids.
However, large amounts of DNA are needed to ligate into a plasmid before cloning can happen. This is why PCR is first used to amplify the positive control DNA using cloning primers with KpnI sites, which differ from the previous qPCR primers. PCR is utilized to amplify enough DNA to ligate and clone into the plasmid. For this perspectives article, the pUC57 plasmid, which has an ampicillin resistance marker, is used. The marker causes bacteria containing pUC57 with the cloned DNA to grow in the presence of ampicillin, and prevent the bacteria that do not take up the plasmid from surviving and growing.
A restriction enzyme cuts, or digests DNA segments at specific sites or codes. For our article, we use the KpnI restriction enzyme. This is the KpnI restriction site:
GGTAC C
C CATGG
The red highlighted the sections where KpnI restriction enzyme digests.
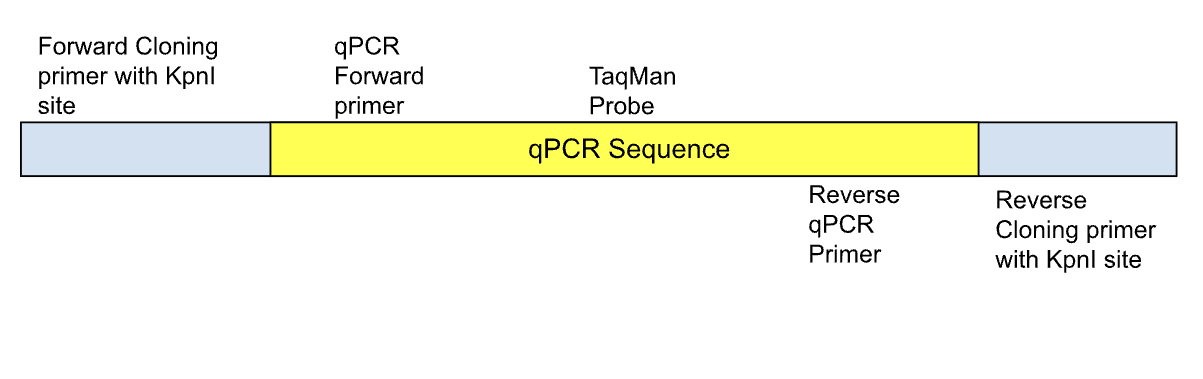
| Figure 7. The qPCR Sequence. This figure represents positive controls that are cloned in pUC57 for each gene: PHOX2B, SHANK3, and MECP2 wild type and mutant. |
|
|---|
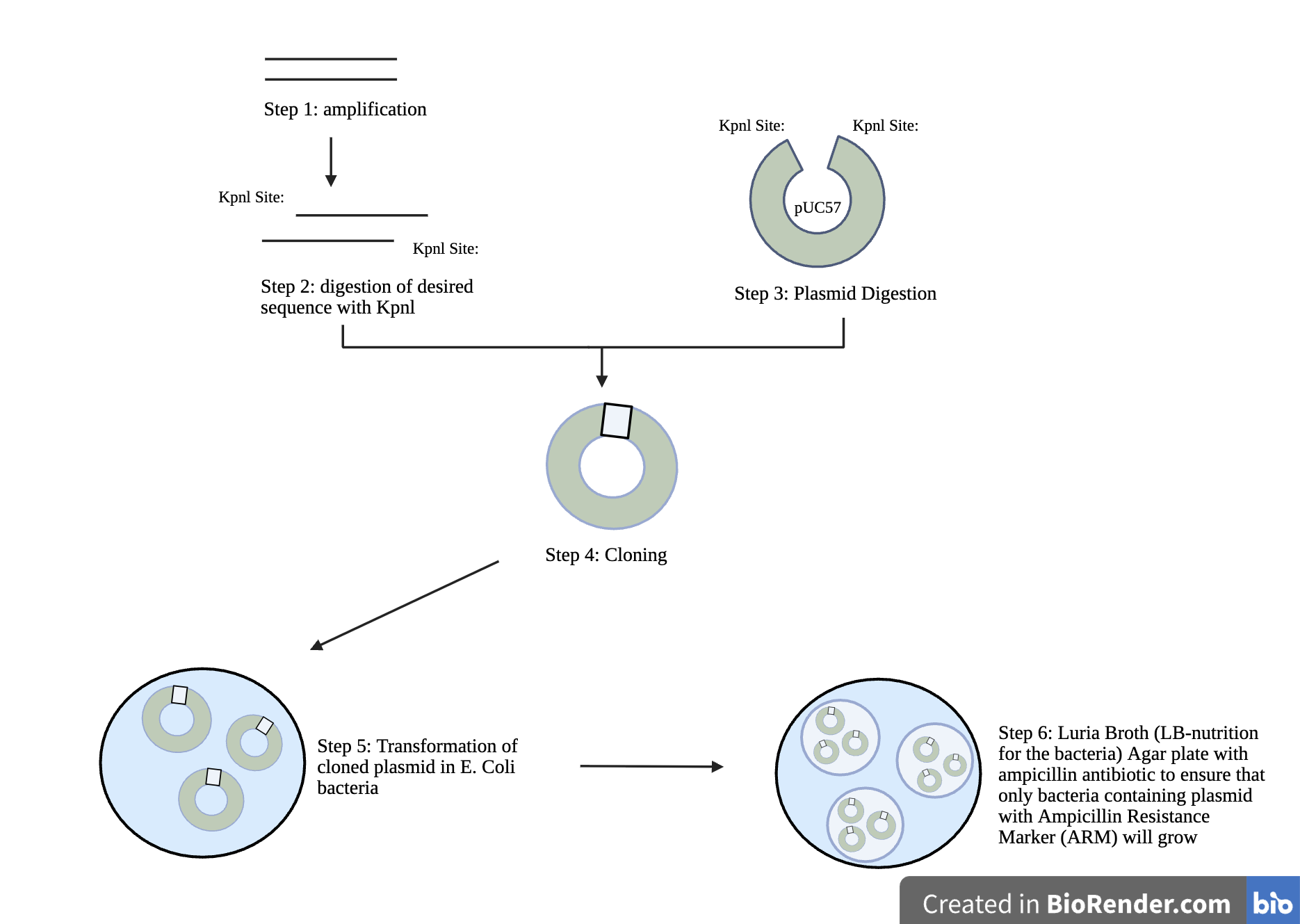
| Figure 8. The Cloning Workflow for Positive Controls. There are five steps necessary to clone positive controls. To begin, the DNA sequence is amplified, then the desired sequence is digested with Kpnl, and next, plasmid digestion occurs. Following step three, cloning occurs, in which the amplified KpnI-digested positive control sequence is mixed with digested plasmid pUC57 from step 3 to form a plasmid clone, which becomes the positive control. The cloned plasmid is then transformed into E. coli bacteria. Lastly, the cloned plasmid from step five is selected on a nutrient plate containing ampicillin to select the bacterial cells that have taken up the plasmid, allowing these bacteria to grow. These positive control bacteria are preserved to supply positive control DNA for the qPCR chips. Plasmid DNA can be extracted from bacterial clones using commercially available kits. |
|
|---|
Positive control DNA is used for the development and validation of our qPCR assay, eliminating the need for biological samples for the development work.
DNA Extraction from Blood
For the development and validation of the chip, the positive and negative control DNAs are used. For the actual diagnostics, the DNA from patient samples is first extracted, quantified, and the approximate amount that gives a similar signal as the controls is determined. This is an additional step. At this stage, a qPCR control is also added for 2 conserved gene regions where a mutation is generally not expected. This control could be the housekeeping gene beta-actin that codes for a cytoskeletal structural protein (NM_001101.5). This is included in later stages of our prototype development after we establish the chip with the three gene targets presented in this perspectives article.
For DNA extraction, commercially available kits will be used, such as QIAGEN QIAamp kit which follow lysis of the cell in the blood sample and binding the extracted DNA to a silica column, followed by washing steps to remove the impurities and PCR inhibitors such as hemoglobin and finally elution of purified DNA in water or a PCR compatible buffer. The purified DNA is quantified to the exact amount needed and added to the qPCR reaction.
Finding the Mutation and Designing Primers and Probes
Step 1: Find the gene mutations using the ClinVar Database https://www.ncbi.nlm.nih.gov/clinvar/
The first step to designing positive control DNA is to determine if genetic mutations are known and associated with the disease. The ClinVar database contains the exact mutations with references to the wild-type sequence. Once we know the mutation, the nucleotide positions that are mutated, and the reference DNA, we can design two probes covering the region of mutation. One probe is specific to the wild-type sequence, and the other probe is specific to the mutated sequence.
Step 2: Find the gene sequence for the wild-type gene using the following database. https://www.ncbi.nlm.nih.gov/nuccore
Once we know the reference for the wild-type sequence from the ClinVar database, we can search the National Library of Medicine (NCBI) database to know the exact sequence of the reference. Once we have the sequence, we can design primers and probes around the region of mutation such that the PCR product is around 100 base pairs.
Step 3: Create a Mutant sequence based on the information from ClinVar
We can manually change the sequence representing the mutant from the information in ClinVar for the mutant control sequence. The mutation for the three genes can be found in the ClinVar database “PHOX2B“[GENE] – ClinVar – NCBI; “MECP2“[GENE] – ClinVar – NCBI; “SHANK3“[GENE] – ClinVar – NCBI.
Step 4: To design the primers and probes, software such as the Thermo Fisher TaqMan Primer Design is used. The primers and probes have an annealing temperature of around 60°C.
When the difference between wild-type and mutant genes is only one base pair, such as in the SHANK3 and MECP2 genes, the probe from the wild-type may bind the mutant sequence weakly, and vice versa, and the cycling temperature has to be optimized to obtain specificity. Similarly, he mutation has to be placed in the middle of the probe to provide specificity. Lastly, differences are detected by looking at the cycling time of PCR, meaning how fast the amplification happens.
For qPCR, the conditions are regulated as follows for the first trial and optimized to improve performance.
- Master mix: TaqMan Universal Master Mix
- Annealing temperature: 60°C
- Cycles: 40
Development of qPCR prototype chip
To advance the development of the qPCR chip for genetic diagnostics of rare diseases, we propose to proceed with Laboratory Validation, Modeling and Optimization, and the development of a prototype chip.
- Laboratory validation of the qPCR reactions in a PCR plate
To begin, primer and probe sets are tested for the respective rare disorders in a PCR plate using commercially available equipment and reagents. This ensures an accurate detection of gene mutations and a reliable amplification of DNA sequences.
- Modeling and optimization
After gathering data from the first runs, primer and probe sequences are refined for accuracy and specificity to the DNA sequences.
- Development of a prototype chip
After validating our qPCR assays in a PCR plate, these reactions are transferred into a chip to simplify routine diagnostics. The qPCR chip has individual reaction wells for numerous rare disease gene targets, thus avoiding the complexities associated with multiplexing. Once the chip is created, a technician simply needs to load the patient DNA sample and run the pre-designed and validated qPCR cycle to screen for numerous rare diseases on the same chip with one sample.
To manufacture our prototype, one of the following companies that offer custom qPCR chip manufacturing services is approached. A sequence of primers and probes, and control DNA are provided.
- Thermo Fisher Scientific – TaqMan OpenArray
- Fluidigm – Microfluidic qPCR Chips
- Qiagen – Custom qPCR Panels
- Bio-Rad – qPCR Arrays
Once loaded, the DNA samples flow through the microfluidic system, and reaction wells of the chip are loaded with exactly 33 nanoliters per well. The chip is then sealed to prevent evaporation, then it is mounted on the chip holder frame of the qPCR machine designed for chips, such as the Applied Biosystems QuantStudio system. This machine is programmed to run qPCR and display results from all 3,072 wells in real time.
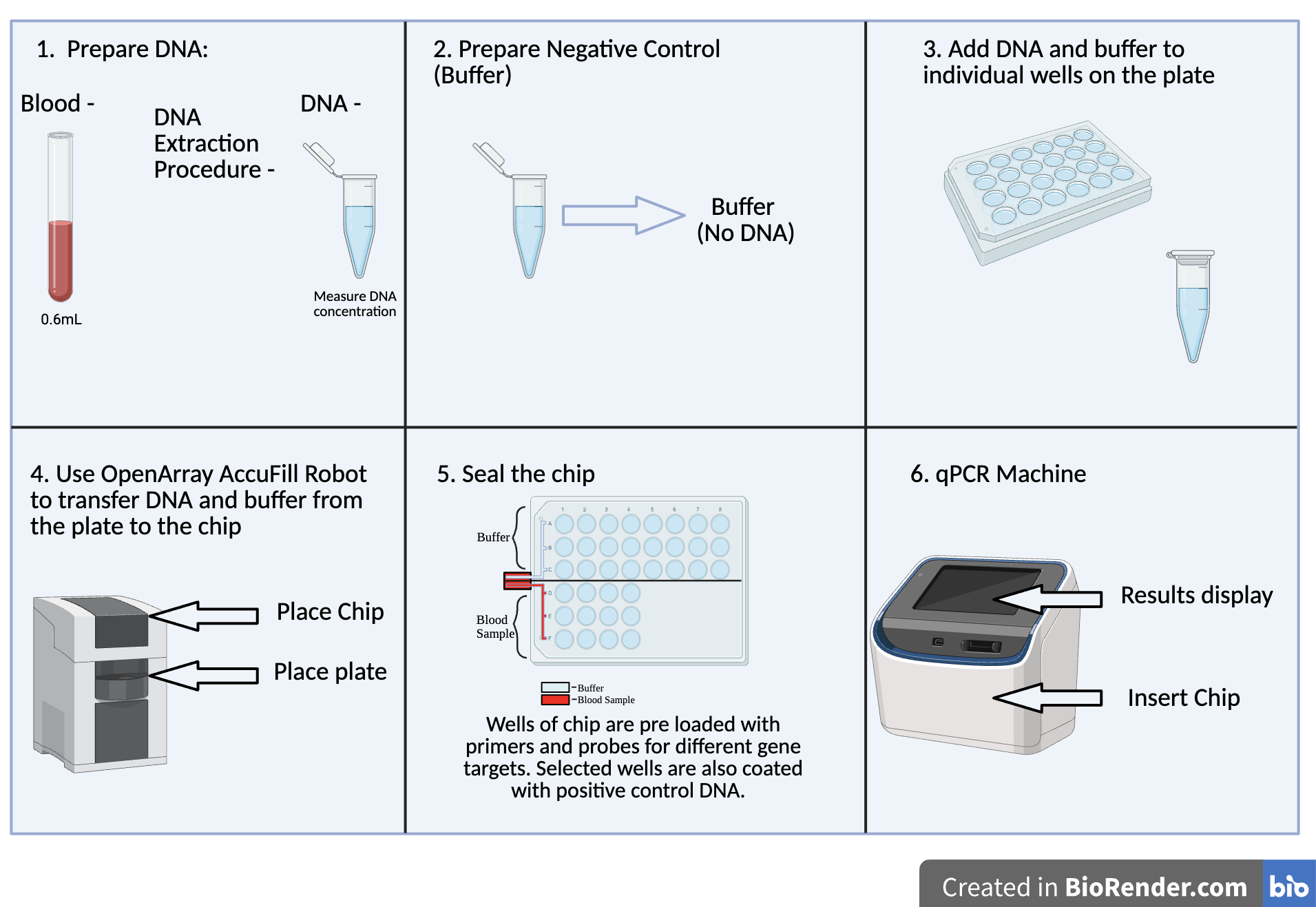
| Figure 9. Development of the qPCR Chip. There are 6 steps by which the qPCR chip is developed. To begin, DNA is extracted from the 0.6 mL blood sample. Following this, the negative control or buffer without DNA is prepared. Afterwards, DNA from the blood sample and the buffer is added to each individual well on the plate. Next, using the OpenArray AccuFill Robot, DNA and the buffer are transferred from the plate onto the chip. The chip is then sealed and inserted into the qPCR machine from which results can be obtained. |
|
|---|
Next Steps
Patient and Clinician Engagement
We will collaborate with CCHS, ASD, and other rare disease patient advocacy groups and medical professionals on outreach programs to explain the clinical utility of the qPCR chip in genetic diagnosis to the general population and medical professionals. Alongside the outreach programs, surveys to assess the likelihood of families, genetic counselors, and clinicians to adopt this technology for early disease screening will be useful in improving the outreach programs.
Expanding qPCR chip for the diagnosis of other Rare Diseases
Once we establish the qPCR for CCHS (PHOX2B gene) and ASD (SHANK3 and MECP2), we can expand the chip to include other rare diseases for which the genetic mutations are well-characterized, such as:
Mitochondrial Diseases: Diseases caused by defects in mitochondria. Mitochondrial Diseases affect body parts including the brain, kidney, and heart (National Institute of Neurological Disorders and Stroke, 2025). Diagnosis of Mitochondrial Diseases can be prolonged and invasive.
Inherited Cancer Syndromes: Conditions such as Lynch syndrome and familial adenomatous polyposis are generally associated with specific genetic mutations. Early genetic screening can permit timely management and coping strategies.
Neuromuscular Disorders: These disorders include Duchenne muscular dystrophy and spinal muscular atrophy, which have well-characterized genetic underpinnings. Rapid genetic testing will expedite any necessary therapeutic intervention.
Traditional symptom-based diagnostics are a lengthy process that has drastic effects on patients due to delayed treatments. qPCR allows for the rapid and simultaneous detection of multiple rare disorders to be screened for at once. This streamlined and accurate diagnostic method will improve outcomes and quality of care for patients.
Potential Pitfalls and Troubleshooting Strategies
While the proposed qPCR chip offers numerous advantages, several potential challenges must be addressed.
Primer-dimer formation and/or non-specific amplification can potentially affect the accuracy or sensitivity of the qPCR reaction. To avoid this and obtain optimal performance, we have taken the following precautions in the design, such as using a TaqMan probe that provides additional specificity, testing with positive control and negative control DNA to ensure specificity. In addition, we will try several reaction conditions to select the optimized assay conditions first in a 96-well plate before ordering the chip. We will repeat selected reaction conditions on the chip to ensure accuracy and sensitivity of the reaction and further optimize as necessary. Furthermore, we will share the primer and probe sequences and target region information in open databases to encourage collaboration, validation, and expansion by the scientific community.
While qPCR offers a real-time and cost-effective method for identifying known genetic mutations associated with rare diseases, clinical management will still require in-depth evaluations, including metabolic assessments, imaging, and symptom-based analysis to guide appropriate therapy.
Author Contributions
A.A. and K.D. equally contributed to the research and writing of the manuscript. A.A. and K.D. worked together to learn and research various topics, including rare diseases. qPCR and TaqMan. Both contributed to the creation of images and to the development of the video used to explain our motivations, prototype, and qPCR chip design.
Acknowledgements
Dr. Lindsey L’Ecuyer and Dr. Vandana Dole made this study possible by not only offering their insights but also dedicating countless hours to passing on their knowledge. Additionally, they both provided essential feedback used to improve the manuscript. We are truly grateful for this opportunity, which would not have been possible without the time and resources of our mentors. While Dr. L’Ecuyer guided us through the writing process, she also aided us in the initial research stages of this project as well as the editing and revising process. She also encouraged us to submit to BioTreks as the teacher advisor at our school’s BioBuilders club. Dr. Dole also played an essential role in the development of the manuscript by introducing us to new topics and ensuring our comprehension of them was complete.
Reference
Breiling, A., Lyko, F. (2015). Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics & Chromatin 8, 24. https://doi.org/10.1186/s13072-015-0016-6
Breining, G. (2017). Rare Diseases Difficult to Diagnose, Cures Hard to Come By. AAMC. https://www.aamc.org/news/rare-diseases-difficult-diagnose-cures-hard-come
CCHS Network. (2025). What is CCHS? https://cchsnetwork.org/what-is-cchs/
Chang, D. F., Gilliam, E. A., Nucho, L. A., Garcia, J., Shevchenko, Y., Zuber, S. M., Squillaro, A. I., Maselli, K. M., Huang, S., Spence, J. R., Grikscheit, T. C. (2021). NH2-terminal deletion of specific phosphorylation sites on PHOX2B disrupts the formation of enteric neurons in vivo. Am J Physiol Gastrointest Liver Physiol. 320(6):G1054-G1066. https://doi.org/10.1152/ajpgi.00073.2021
Cleveland Clinic. (2022). Parasympathetic Nervous System (PSNS). https://my.clevelandclinic.org/health/body/23266-parasympathetic-nervous-system-psns
Cleveland Clinic. (2025). Congenital Central Hypoventilation Syndrome (CCHS). https://my.clevelandclinic.org/health/diseases/24816-congenital-central-hypoventilation-syndrome
Dana Foundation. (2023). Neurotransmission: The Synapse. https://dana.org/resources/neurotransmission-the-synapse/
Du, Q., Luu, P.-L., Stirzaker, C., & Clark, S. J. (2015). Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics, 7(6), 1051-1073. https://doi.org/10.2217/epi.15.39
Dudoignon, B., Maruani, A., Delorme, R., Patout, M., Fefeu, M., Ellul, P., Bokov, P., & Delclaux, C. (2024). Autism spectrum disorder in young patients with congenital central hypoventilation syndrome: role of the autonomic nervous system dysfunction. Orphanet Journal of Rare Diseases, 19(1), 249. https://doi.org/10.1186/s13023-024-03257-z
Hering, H. and Sheng, M. (2001). Dendritic spines : structure, dynamics and regulation. Nature Reviews Neuroscience, 2, 880-888. https://doi.org/10.1038/35104061
Mark, M., Rijli, F. M., & Chambon, P. (1997). Homeobox Genes in Embryogenesis and Pathogenesis. Pediatric Research, 42, 421-429. https://doi.org/10.1203/00006450-199710000-00001
MedlinePlus. (2025). What is the cost of genetic testing, and how long does it take to get the results? https://medlineplus.gov/genetics/understanding/testing/costresults/
MIT News Office. (2018). Electrical properties of dendrites help explain our brain’s unique computing power. MIT News. https://news.mit.edu/2018/dendrites-explain-brains-computing-power-1018
Moore, L. D., Le, T., & Fan, G. (2012). DNA Methylation and Its Basic Function. Neuropsychopharmacology, 38, 23-38. https://doi.org/10.1038/npp.2012.112
Nagarajan, R. P., Hogart, A. R., Gwye, Y., Martin, M. R., & LaSalle, J. M. (2007). Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics, 1(4), 1-11. https://doi.org/10.4161/epi.1.4.3514
National Institute of Environmental Health Services. (2025). Autism. https://www.niehs.nih.gov/health/topics/conditions/autism
National Institute of Neurological Disorders and Stroke. (2025). Mitochondrial Disorders. https://www.ninds.nih.gov/health-information/disorders/mitochondrial-disorders
National Library of Medicine. (2025a). PHOX2B paired like homeobox 2B [ Homo sapiens (human) ]. https://www.ncbi.nlm.nih.gov/gene/8929
National Library of Medicine. (2025b). SHANK3 SH3 and multiple ankyrin repeat domains 3 [ Homo sapiens (human) ]. https://www.ncbi.nlm.nih.gov/gene/85358
National Library of Medicine. (2025c). MECP2 Methyl-CpG Binding Protein 2 [ Homo sapiens (human) ]. https://www.ncbi.nlm.nih.gov/gene/4204
Phillips, C., Parkinson, A., Namsrai, T., Chalmers, A., Dews, C., Gregory, D., Kelly, E., Lowe, C., & Desborough, J. (2024). Time to diagnosis for a rare disease: managing medical uncertainty. A qualitative study. Orphanet journal of rare diseases, 19(1), 297. https://doi.org/10.1186/s13023-024-03319-2
Sarowar, T., & Grabrucker, A. M. (2016). Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural plasticity, 2016, 8051861. https://doi.org/10.1155/2016/8051861
Waxenbaum, J. A., Reddy, V., Varacallo, M. A. (2023). Anatomy, Autonomic Nervous System. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK539845
Wen, Z., Cheng, T.-L., Li, G.-Z., Sun, S.-B., Yu, S.-Y., Zhang, Y., Du, Y.-S., & Qiu, Z. (2017). Identification of autism-related MECP2 mutations by whole-exome sequencing and functional validation. Molecular Autism, 3, 8-43. https://doi.org/10.1186/s13229-017-0157-5