Ines Belza Garcia, Dylan Chang, Kristin Chang, Bernie Chen, Guan Chen, Alexander Cheng, Sienna Chien, Rowan Delgado-Marquez, Minna Hang, Sabrina Hong, Andrew Hsiao, Sean Hsu, Kaitlyn Hu, Evelyn Huang, Laura Hwa, Sean Kao, Yvonne Kuo, Alan Lee, Jane Lee, Jason Leu, Jeffrey Li, Andreea Liao, Easton Liaw, Clarence Lin, Emily Lu, Lindsey Nien, Ethan Su, Mabel Tan, Wu Han Toh, Justin Wang, Christine Wong, and Annabelle Wu
Taipei American School, Taipei, Taiwan
Reviewed on 7 May 2022; Accepted on 25 June 2022; Published on 15 October 2022
With help from the 2022 BioTreks Production Team.
Blood transfusions are an integral component of healthcare, but the availability of viable blood is limited by patient-donor blood type specificity. This contributes to seasonal shortages as well as shortages worldwide, especially in developing countries and during pandemics or natural disasters. Attempts to increase blood supply with commercial incentives have raised ethical concerns, and current proposed artificial blood substitutes are unable to fully replicate the function of native red blood cells (RBCs). In this study, we explore the potential strategy of alleviating blood shortages through enzymatic conversion of A, B, and AB blood types to O types. This process eliminates ABO patient donor incompatibility, increasing the supply of universal donor blood. We selected three glycoside hydrolases acting as molecular scissors to cleave terminal residues on A and B RBC surface antigens and catalyze the conversion process. We recombinantly expressed these enzymes in BL21(DE3) Escherichia coli cells and purified them through nickel ion affinity chromatography. We used a combination of colorimetric substrate assays, thin-layer chromatography, and mass spectrometry to evaluate enzyme functionality. We modeled enzyme efficiency using Michaelis–Menten kinetics. We demonstrated partial enzymatic A-to-O blood type conversion on porcine red blood cells with slide agglutination tests. Ultimately, we envision a modular, column-based, two-step blood conversion kit housing our recombinant enzymes that can be implemented in blood banks and processing centers, making universal donor blood more accessible to all.
Keywords: ABO blood group system, blood shortage, glycoside hydrolase, porcine red blood cells, antigen cleavage
Authors are listed in alphabetical order. Jude Clapper and Jonathan Hsu mentored the group. Please direct all correspondence to 22enocht@students.tas.tw, 22yvonnek@students.tas.tw, hsujo@tas.edu.tw, and clapperj@tas.edu.tw.







Background
Blood transfusions are essential in healthcare, as they are used to replace blood that is lost or provide blood when one’s body lacks normal production. Transfusions are needed in both routine and emergency situations, such as after an injury, during surgery, and for people with blood-related disorders (NIH, 2022). Transfusions save millions of lives each year, drastically improving patient life expectancy and quality of life (WHO, 2010).
The availability of blood for transfusions is regulated by the supply and demand of safe, transfusable blood. Because blood products can only be derived from healthy donors, their supply is limited by public willingness to donate. In the past, the WHO has estimated that a minimum donation rate of 1% of the total population is required to satisfy the most basic requirements for blood (WHO, 2010). Although this is a low percentage of the world population, national and international organizations have often struggled to meet these targets.
A blood shortage occurs when the immediate blood supply fails to meet local demand. They are especially prevalent in aging populations where increasing medical care is required, in developing countries where chronic blood shortages are common due to limited health services and access to safe blood, and in developed countries where complex blood-intensive medical procedures are more common (WHO, 2010). Fluctuations causing seasonal and periodic shortages are also regularly observed since the blood market operates without market prices and with a short shelf-life (Slonim et al., 2014). Many regions have also experienced concerning long-term trends like decreasing donor rates among younger populations that further contribute to low supply (Lemmens et al., 2005).
The COVID-19 pandemic, in particular, has exacerbated the blood shortage issue worldwide, as it has caused decreased blood donations and put a strain on blood bank staff (Stanworth et al., 2020). Using the authors’ home residence of Taiwan and its 1st-wave surge in COVID-19 cases as an example, it has been reported that Taiwan’s blood supply has fallen to the lowest in 20 years (Taiwan News, 2021). Significant drops in donations have also been noted in the African region, and the American Red Cross reported a 10% decline in donations in the United States since the pandemic (Loua et al., 2021; American Red Cross, 2022). The significance of blood transfusions calls for more robust measures to ensure national blood supply levels can satisfy demand.
Current attempts to alleviate the blood shortage include incentivization and using artificial substitutes. Some organizations have turned towards incentives such as points, gifts, or other redeemable benefits to attract donors and increase the donor pool. However, ethical issues arise with this approach when incentivization is taken a step further to include paid or commercialized donors that donate blood regularly to a blood bank for an agreed fee or sell their blood to banks or patients’ families. Blood from these donors may not be as reliable or safe, as studies have reported that paid donors have the highest prevalence of transfusion-transmissible infections, as they may be motivated by financial factors to withhold information about their conditions (WHO, 2010). Commercializing blood further raises concerns as blood donations should respect an individual’s rights to their own body, but a commercialized market may give way to exploitation (WHO, 2010).
Artificial blood substitutes are synthetically constructed to transport oxygen and carbon dioxide throughout the body. The ideal artificial blood product is safe and compatible with all blood types of the human body, is able to be sterilized to remove disease-causing viruses and microorganisms, and can be stored for over a year (Sarkar, 2008). There are currently two types of RBC substitutes, perfluorocarbon-based and hemoglobin-based, that differ in the way they carry oxygen (Moradi et al., 2016). However, problems such as solubility and stability need to be overcome for such products to be feasible. Scientists still need to learn more about how natural RBCs function to produce a substitute with fewer side effects, increased oxygen-carrying ability, and longer life spans in the human body (Artificial Blood, n.d.). As such, utilizing the existing blood supply with a different approach can act as a potential solution.
Armed with knowledge of these limitations, we sought an approach that would both preserve the altruistic nature of donations and the native function of red blood cells. In recent years, the notion of enzymatic blood-type conversion to eliminate transfusion compatibility has been of renewed interest (Rahfeld & Withers, 2020). The membranes of Red Blood Cells (RBCs) contain protein and sugar antigens that, if recognized as foreign, elicit an adverse immune response in the human body (Dean, 2005). The presence of different antigens on different individuals’ RBCs is responsible for patient-donor incompatibility.
While RBCs possess many different antigens, the ABO blood group system holds the greatest clinical significance in transfusions as its antigens are the most immunogenic (Mitra et al., 2014). Similarities and differences in the basic structures of ABO blood group antigens are depicted (Fig 1). The H-antigen is a precursor of A and B antigens and is found on nearly all RBCs. The A antigen is found on type A RBCs, and the B antigen is only found on type B RBCs, while the enzymes required to create A and B antigens are codominantly expressed in AB blood types. Because type O blood lacks foreign A and B antigens, it is considered the “universal” blood type and is often valuable for emergency transfusions and immune deficient infants, as it can be given to all patients with an Rh-positive blood type (Blood Types Explained – A, B, AB and O | Red Cross Blood Services, n.d.). Because A and B antigens on type A, B, and AB blood differ from the H-antigen only in the addition of a single sugar group, using enzymes to catalyze the cleavage of the terminal linkages on the antigens can effectively convert A and B blood to O type blood (Rahfeld and Withers, 2020).
Using a model we created to simulate the weekly supply and demand of blood using historical data from the Taiwan Blood Services Foundation, we found that over a simulated period of 800,000 years, using 10% fluctuation magnitudes, there would be a shortage due to incompatibility 9.98% of the time, which amounts of over one month each year. We speculate that even in a country like Taiwan, with one of the highest donation rates in the world, there are considerable applications for plausible blood type conversion technology. The complete code used to implement our model can be assessed on GitHub (https://github.com/igemsoftware2021/TAS_Taipei/releases/tag/1.0.0).
In this study, we aim to consolidate and expand current work on enzymatic blood type conversion. Through a systematic literature search, we identified three glycoside hydrolases isolated from bacterium capable of cleaving the appropriate sites on A and B antigens to catalyze successful blood type conversion: α-N-acetylgalactosaminidase (NAGA) from Elizabethkingia meningoseptica that cleaves the terminal residue on A-antigens, α-galactosidase (α-gal) Bacteroides fragilis that cleaves the terminal residue on B-antigens, and endo-1,4-β-galactosidase (endo-β-gal) from Streptococcus pneumoniae that cleaves A and B antigen trisaccharides (Rahfeld & Withers, 2020) (Kwan et al., 2015) (Liu et al., 2007) (Liu et al., 2008) (Higgins et al., 2009). These enzymes were chosen based on their ability to cleave their respective blood group antigens at neutral pH and room temperature, properties that enable direct conversion of RBCs in blood and eliminate the need for incubation (Table 1). By using these enzymes in combination, we hope to achieve a more thorough conversion than using one enzyme alone.
Table 1. Properties of selected enzymes. (Rahfeld & Withers, 2020; Kwan et al., 2015; Liu et al., 2007; Liu et al., 2008; Higgins et al., 2009).
Note. – The sequence for endo-β-gal used in this study was modified according to the directed evolution performed by Kwan et al. (2015). The properties displayed in this table, however, reflect that of the wild-type enzyme.
We aim to recombinantly produce these enzymes through high-level expression in BL21(DE3) E. coli. Then, by purification through nickel ion affinity chromatography, we evaluate the functionality of the enzymes through a combination of tests that include mass spectrometry, which separates compounds based on molecular mass, thin layer chromatography (TLC), which separates based on polarity, and colorimetric tests, which use substrates that change color upon successful cleavage. To evaluate the ability of our enzymes to cleave actual blood antigens on the surface of RBCs, we perform agglutination tests with porcine red blood cells (pRBCs). pRBCs share several similar characteristics with human red blood cells and possess antigens that closely mimic the A, B, and H antigens of human RBCs (Fig 2) (Smood et al., 2019; Cooling, 2015). Because of these similarities, porcine blood is the most promising candidate for xenotransfusions (transfusons that take place across species) (Long et al., 2009). The similar properties between pRBCs and human RBCs also make it an ideal substitute for human RBCs in our experiments, which allows us to avoid the safety questions that arise when working with human blood. Since the A-O antigen system exists within pRBCs, the activity of NAGA can be evaluated through enzymatic reactions followed by agglutination assays with type A and O porcine blood (Mujahid & Dickert, 2015). In theory, successful cleavage of the A antigen in porcine RBC should eliminate observation of agglutination when human anti-A serum is added to the treated type A porcine blood.
To improve the feasibility of using our enzymes for blood type conversion, we propose several procedural modifications, informed by consultations with blood banks and hematologists. First, we propose immobilizing our enzymes on beads housed in sealed columns. By doing so, we lower the chance of enzymes falling into the solution during conversion. Using columns further will allow the conversion process to occur in an airtight setting. Next, we introduce post-conversion processing steps aimed at preparing the converted product to be safe for transfusion. We will do this through a combination of centrifugation, filtration, and antibody agglutination to purify converted RBCs from unconverted RBCs and various waste products. In its entirety, we envision this system being implemented in the blood processing step of the supply chain and activated when there is a shortage of O-type blood.
Materials and methods
Sequence retrieval and plasmid design
The amino acid sequences of α-galactosidase (α-gal), α-N-acetylgalactosaminidase (NAGA), and endo-1,4-β-galactosidase (endo-β-gal) were retrieved from the UniProt database (https://www.uniprot.org/), with accession numbers of Q5L7M8, A2AWV6, and A0A0H2UKY3, respectively. Certain amino acids of the endo-β-gal sequence were selectively modified according to a structural directed evolution performed by Kwan et al., who reported an increase in its activity towards more antigen subtypes (Kwan et al., 2015).
Following reverse translation of the sequences, gene constructs were created by attaching a T7 promoter derived from the T7 phage and a strong ribosome binding site (http://parts.igem.org/Part:BBa_K525998) upstream of the enzyme sequence. An N-terminal or C-terminal 6x Histidine tag was added for protein purification purposes through a flexible Glycine-Serine linker. A double terminator (http://parts.igem.org/Part:BBa_B0015) downstream of the open reading frame was also added. The gene sequences were codon optimized for expression in E. coli using the genscript tool (https://www.genscript.com/). All composite genes were synthesized through Integrated DNA Technologies. Sequences for the plasmids used in this study are available through the Registry of Standard Biological Parts (Fig 3).
Protein expression and purification
The synthesized plasmids were transformed into BL21 (DE3) E. coli at 37°C overnight, diluted, and grown to OD600 0.5~0.6 at 37°C. Cultures were induced for expression with 0.5 mM IPTG and allowed to grow overnight at room temperature. Cells were harvested by centrifugation, and cell pellets were lysed through xTractor Lysis Buffer (Takara, Japan) supplemented with 20 mM imidazole. Purification of our histidine-tagged proteins was done using Ni sepharose affinity chromatography. SDS-PAGE was utilized to confirm the sizes of purified proteins. Purified enzymes were transferred to a reaction condition solution (hereinafter referred to as sodium phosphate buffer) consisting of 20mM NaH2PO4, 130mM NaCl, and 1mM DTT adjusted to pH 7 using NaOH through buffer exchange dialysis at 4°C (Hsieh et al., 2003).
Functional Tests
For enzymes that were successfully expressed, their functionality was verified using a series of experiments (Fig 4).
Colorimetric substrates
Colorimetric substrates for our NAGA and α-gal enzymes were purchased from Sigma Aldrich (Fig 5). These substrates contain a 4-nitrophenol leaving group, which turns yellow upon successful cleavage in solution. The concentration of 4-nitrophenol can be quantified at absorbance 405 nm using a 96 well plate assay (Held, 2007).
Small-scale colorimetric tests were conducted to verify the function of the enzymes. 50 μL of 10 mM substrate, 10 μL of enzyme, 30 μL of water, and 10 μL of 10x Glycobuffer 1 (50 mM CaCl2, 500 mM sodium acetate, pH 5.5), a buffer recommended by New England Biolabs to ensure optimal enzyme activity (New England Biolabs, n.d.), was added to each well (Table 2). A non-specific substrate and nitrophenol reaction product were used as negative and positive controls, respectively. Following 2 hours of reaction at room temperature, well contents were diluted in 1.9 mL of water, and absorbance readings were taken at 405 nm with a CT-2400 Spectrophotometer (ChromTech, USA).
Table 2. Buffers and solutions used in substrate and control wells for colorimetric enzyme experiments.
To extrapolate enzyme-substrate cleavage efficiency from experimental data, Michaelis-Menten Kinetics was used for modeling. Michaelis-Menten Kinetics helps determine the relationship between enzyme concentration and reaction rate (Lee et al., 2020). Ultimately, the model utilized the following relationship:
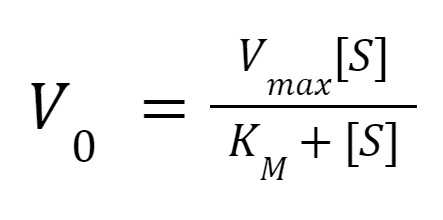
The substrate concentration, [S], is the independent variable. V0, the initial velocity/rate of reaction, is the dependent variable. Vmax denotes the maximum reaction rate achieved by increasing substrate concentration. Km denotes the substrate concentration at which the initial reaction rate is ½ the maximum. Vmax changes with different enzyme concentrations, but Km stays constant. Taking the inverse of the relationship above, the following can be obtained:
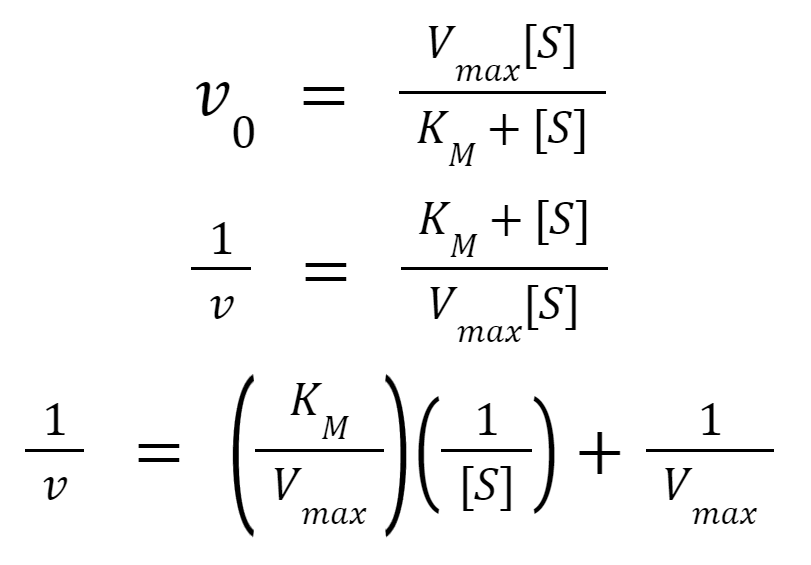
As a result, a linear regression model can be applied to experimental data to determine the Km and Vmax. This is also called the Lineweaver Burk plot in the context of Michaelis Menten Kinetics (Lee et al., 2020). Since Kₘ remains constant across different enzyme concentrations, the initial reaction rates at different substrate concentrations (equal to Kₘ) can be measured with enzyme concentrations to obtain the ½ Vmax for any given enzyme concentration. We can then multiply by 2 to get the Vmax for the given enzyme concentration. A linear relationship between enzyme concentration and Vmax (maximum reaction rate) can be determined with a y-intercept of 0 (Lee et al., 2020).
To quantify the activity of the enzymes, enzyme reactions were performed at various substrate concentrations. The absorbance was measured at different time intervals over a constant time period with a multimode plate reader (Tecan, Switzerland). Results were averaged from at least 3 independent trials from the colorimetric assay to calculate Michaelis–Menten constants for enzyme efficiency. To quantitatively determine enzyme efficiency, a conversion relationship between absorbance and concentration must be determined. Therefore, a calibration curve for absorbance units versus concentration of 4-Nitrophenol product was plotted. The hyperbolic relationship between substrate concentration and reaction rate for both α-galactosidase and α-N-acetylgalactosaminidase was then calculated and graphed. This provided the Km values for both enzymes. With substrate concentrations equal to Km, enzyme concentrations were varied to determine the linear relationship between enzyme concentration and reaction rate.
Thin layer chromatography
Melibiose was used as a substrate for α-gal, followed by detection through normal-phase thin-layer chromatography (TLC), which utilizes the competition of the solute and mobile phase for binding sites to separate compounds based on polarity. α-gal can cleave the bonds in melibiose, causing the dissolution of melibiose into its constituent monosaccharides, glucose and galactose (Fig 6).
To create the reference standards, controls of melibiose, as well as glucose and galactose were spotted onto silica plastic-backed TLC plates (Silicycle, Canada), then ran with a mobile phase of ethyl acetate:ethanol:acetic acid:boric acid (5:2:1:1, v/v/v/v). The plate was visualized with a spraying reagent consisting of sulfuric acid:ethanol (1:1, v/v), followed by heating at 140°C for 5 minutes. Bands were observed both in ambient light and at 365 nm, and hRf values were measured as the solute front divided by the solvent front multiplied by 100. Based on our experiments with three independent trials, we determined that glucose and galactose migrate significantly further up the plate compared to melibiose, with hRf values of 55.9 ± 2.8 for glucose and galactose, and 33.1 ± 1.4 for melibiose (Fig 7).
With this TLC detection method in place, we tested the functionality of α-gal. 10 μL of 10 mg/mL melibiose was incubated with 5 μL of enzyme or water (negative control) and 5 μL of 0.1 M citrate-phosphate buffer (pH 5.8) at room temperature for a total reaction time of 2 hours. The control and enzyme reaction sample was then spotted on TLC plates and developed to see if successful hydrolysis would occur and if glucose and galactose would be present.
Mass spectrometry
The enzymes’ specificity was further tested by using actual antigen trisaccharides as substrates. These trisaccharides contain three monosaccharides that have identical chemical structures to the A and B antigens found on RBCs. A reaction was carried out of the enzyme and its corresponding trisaccharide dissolved in 1X GlycoBuffer 1 (New England Biolabs, n.d.), deionized water, and purified BSA for 1 hour at 37°C. A-antigen and B-antigen trisaccharides were kindly provided by Professor Dr. Todd Lowary from the Institute of Biological Chemistry at Academic Sinica (Meloncelli & Lowary, 2010). For each enzyme, a non-specific trisaccharide served as a negative control and the trisaccharide specific to the enzyme served as the experimental unit (Table 3).
Table 3. Experimental setup for enzyme-antigen trisaccharide tests
After the reaction, the reaction solution was passed through C18 columns to minimize impurities from the reaction solution. The flow-through solution was analyzed with a mass spectrometer to measure peaks in molar mass to determine the compounds in the reaction solution. The original molar mass of the A-antigen is 713.35 g/mol (Meloncelli & Lowary, 2010); therefore, after cleavage, two fragments with molar masses of 510.27 g/mol and 203.08 g/mol are expected to form. The original molar mass of the B-antigen is 672.32 g/mol (Meloncelli & Lowary, 2010); therefore, after cleavage, two fragments with molar masses of 510.27g/mol and 162.05 g/mol are expected to form (Fig 8).
Porcine blood agglutination tests
Since the A-O antigen system exists within porcine red blood cells, the activity of NAGA can be evaluated through enzymatic reactions followed by agglutination assays with type A and O porcine blood (Mujahid & Dickert, 2015). In theory, successful cleavage of the A antigen in porcine RBC should eliminate agglutination when human anti-A serum is added to the treated type A porcine blood.
Porcine blood was sourced from a local farm in Hsinchu, a county in Taiwan, specializing in breeding pigs for scientific research purposes (https://www.atri.org.tw/). To determine the blood type of the received porcine blood, initial slide agglutination was performed with the addition of several drops of human anti-A serum to the porcine blood (Mujahid & Dickert, 2015). Previous studies have confirmed that porcine blood agglutinates well with human Anti-A antibodies, and thus should show visible clumps if the A antigen is present (Smith et al., 2006). Our results demonstrated clear clumps in some samples, and no agglutination in others (Fig 9). This difference was used to blood type our pRBCs either as A type or O type.
To test if our NAGA enzyme works on type A pRBCs, slide agglutination was used as a qualitative test to visualize our results. Purified NAGA, previously dissolved in 0.1M sodium phosphate buffer, was dialyzed in 1X PBS to limit harm to pRBCs. Following treatment with the NAGA enzyme, A-type porcine blood should no longer agglutinate upon the addition of anti-A antibodies. Non-enzyme treated type A porcine blood should continue to agglutinate as expected after the addition of anti-A serum. After enzyme treatment for 2 hours, human anti-A serum was added to the blood sample. Agglutination was visualized using 3 different qualitative methods. The blood samples and antibody mixture were spread on standard blood typing slides to observe for macroscopic clumps under a microscope slide at 1x and under a light microscope at 20X magnification (Nikon H550S, Japan).
Enzyme immobilization and post-conversion processing
Based on consultation with hematologists, feedback from blood banks, experimental data, and literature research, a prototype blood type conversion “kit” was designed to first mediate the enzymatic conversion process then purify converted RBCs in preparation for transfusion. The prototype hardware was visualized through Computer Aided Design (CAD) with Solidworks.
Laboratory and environmental safety
We work in a Biosafety Level 1 laboratory, and accordingly, used microbes that posed little to no risk to healthy individuals. Hazardous chemicals and solutions were used at a minimal levelin well-ventilated areas such as fume hoods. Rather than use EtBr for gel electrophoresis, a safer nucleic acid stain called Seeing Safe DNA Dye was used. All chemicals and solutions were treated with due respect, care, and caution. MSDS are stored on our lab computers and in a folder on the lab technician’s desk.
During our experiments, we worked with a safe and common lab strain, E. coli BL21(DE3), and followed safety rules set by our lab instructor. We genetically engineered bacteria to produce our desired enzymes. If bacteria were to escape the lab, they would pose a low risk to the surrounding community. After use, all bacterial wastes were either autoclaved or bleached to kill microbes before disposal. Rather than use human RBCs for testing our enzymes, porcine blood was used to provide a safer alternative for human RBCs. Blood was sourced from a farm in Hsinchu, a county in Taiwan, specializing in breeding pigs for scientific research purposes. With a reliable source and robust handling and screening procedures, our experiments were conducted in a safe manner, and all porcine blood was bleached to kill live cells before disposal. While performing thin layer chromatography, several strong acids were used to create solvents and visualizing reagents, including hydrochloric acid, sulfuric acid, and boric acid. To ensure safety, all solutions and tests were performed in the chemical fume hood while wearing personal protective equipment: latex gloves, goggles, and lab coats. Finally, a thorough clean-up procedure was used. All bacterial liquid wastes were bleached, and used pipette tips were autoclaved before disposal. Other lab wastes were carefully packaged and then sent to a disposal facility to ensure no hazardous materials were mismanaged.
Results
SDS-PAGE confirms successful purification of recombinant α-N-acetylgalactosaminidase and α-galactosidase.
Our results show histidine-α-gal and NAGA-histidine migrating to the expected sizes of 69.7 kDa and 51.7 kDa, respectively (Figs 10-11). A clear band was observed for NAGA, while a faint band was observed for α-gal. Despite our best efforts, we were unable to express or purify histidine-endo-β-gal.
Functional tests yield corroborating results confirming enzyme activity and specificity
Colorimetric substrates
Solutions with colorimetric substrates turned yellow only upon addition of the intended enzyme, demonstrating both the functionality and specificity of the α-gal and NAGA enzyme (Fig 12). When non-specific enzymes were used in the substrate solution, the absorbance did not change, demonstrating that no cleavage occurred. When the appropriate enzyme was added, the solution turned visibly yellow, and a high absorbance was recorded at 405 nm. However, it is interesting to note that these absorbance values were even higher than that of the positive control, suggesting there may have been a mistake in the calibration of the concentration of positive controls.
Thin layer chromatography
When the reaction product of melibiose and α-gal were run on a TLC plate, partial cleavage was observed (Fig 13). Compared to the control of melibiose treated with water, which only displayed one band corresponding to that of melibiose, the reaction product had two visible bands. The top band had a hRf value corresponding approximately to that of glucose and galactose, indicating the enzyme had successfully catalyzed melibiose into its monosaccharide constituents. However, this reaction was not complete, as the original band of melibiose remained present in the product.
Mass spectrometry
Results obtained from the mass spectrometer further confirm the functionality of both α-gal and NAGA. For α-gal, the experimental unit shows the presence of the cleaved fragment from the reaction with the B-antigen trisaccharide, indicated by the peak at 533g/mol in the mass spectrum (Fig 14a). The negative control confirms the specificity of α-gal, as the A-antigen trisaccharide was not cleaved, indicated by the peak at 736g/mol (Fig 15b). For NAGA, the experimental unit shows the presence of the cleaved fragment from the reaction with A-antigen trisaccharide, indicated by the peak at 533g/mol in the mass spectrum (Fig 15c). The negative control confirms the specificity of NAGA, as the B-antigen trisaccharide was not cleaved, as indicated by the peak at 695g/mol (Fig 15d). Throughout all readings of the mass spectrum, the molar mass of the peaks increased by 23 g/mol due to a sodium adduct (Kruve et al., 2013). Unrelated peaks/background noise can be attributed to compounds present in the buffer solution.
Michaelis–Menten kinetics for enzymes were calculated based on colorimetric tests.
To correlate the absorbance units obtained from colorimetric substrate tests with concentrations of the 4-Nitrophenol colorimetric compound, a linear calibration regression was conducted (Fig 15). The linear equation obtained was then used to convert from absorbance units to mM of 4-Nitrophenol.
To quantify the activity of the enzymes, enzyme reactions at various substrate concentrations [S] were performed, and absorbance at initial intervals was measured. The slope of the initial absorbance measurements yielded the initial reaction rates Vi. At least 3 independent trials for each enzyme were used to graph and calculate Michaelis-Menten constants (Fig 16-17). As detailed in the Methods section, experiments were conducted to determine the linear relationship between enzyme concentration and maximum reaction rate (Vmax). With substrate concentration equal to Km and varying enzyme concentrations, the resulting reaction rate (½ Vmax) was multiplied by 2 to obtain the enzyme concentration versus maximum reaction rate (Vmax) relationship. With substrates always in excess, these relationships (Equations 1 and 2) help with determining the duration of reaction time and concentration of enzymes suitable for blood type conversion.
y = 1.6775x
Equation 1. The linear function for α-galactosidase concentration (mM) and Vmax (mM/min), with enzyme concentration being the independent variable and reaction rate being the dependent variable
y = 0.3428x
Equation 2. The linear function for α-N-acetylgalactosaminidase concentration (mM) and Vmax (mM/min), with enzyme concentration being the independent variable and reaction rate being the dependent variable
Porcine blood slide agglutination tests demonstrate successful enzymatic A-to-O blood type conversion.
Following enzyme treatment for 2 hours and the addition of human anti-A serum to the porcine blood sample, agglutination was visualized using 3 different qualitative methods (Fig 18). First, the blood and antibody mixture was spread on standard blood typing slides to observe for macroscopic clumps. Our results showed that enzyme-treated blood displayed fewer clumps compared to PBS-treated blood (control), indicating less agglutination and thus suggesting successful blood type conversion. The mixtures were then observed under a light microscope at 20X magnification. Larger and more RBC clumps in PBS control samples were observed compared to enzyme-treated samples. However, enzyme-treated samples still displayed more clumping than O-type blood, indicating cleavage was incomplete. The corroborative results from the macroscopic slide agglutination test and observation of RBC behavior under the light microscope provide convincing evidence of successful enzymatic cleavage by NAGA, demonstrating a proof of concept for our project.
CAD model depicts blood type conversion kit design for implementation in processing centers.
A two-step blood type conversion kit was designed to 1) mediate the enzymatic conversion process and 2) purify converted RBCs in preparation for transfusion. Prior to enzymatic conversion, donated whole blood will be washed in 0.9% NaCl to remove the plasma and buffy layer coat, a process that has already been implemented into existing procedures at blood donation/processing centers (Souid et al., 2009). A previous study utilized centrifugation settings of 3500 rpm for 5 minutes to isolate RBCs from the solution (Long et al., 2009). Figure 19 shows the components involved in our proposed blood type conversion process.
Enzymes are immobilized on Nickel sepharose beads (see 1, Fig 19) to minimize the chance of enzymes falling into solution but maximize the surface area of enzyme and RBC contact. Using Nickel sepharose beads streamlines the construction process of our kit, as enzymes will already be immobilized on these beads for affinity chromatography during protein purification, saving the additional steps of eluting enzymes from the beads and immobilizing them on another surface. This will also increase the yield of enzymes.
To address concerns over the adverse health effects of beads being accidentally transfused into patients, leukocyte reduction filters (see 2, Fig 19) were added that filter RBCs but trap the nickel sepharose beads (Mizuno, 2013). These filters are currently implemented in many blood processing procedures. The leukodepletion filters will be attached in columns, where they will sit on a layer of biocompatible mesh provided for structural support (see 3, Fig 19). Prior to usage, beads with the appropriate immobilized enzymes will be added to the columns. These columns will be connected in a series of 3, and blood will be passed from the blood bag to the columns with PVC biocompatible tubing (see 4, Fig 19), then through the columns with pressurized flow at a constant rate; the flow speed will thus be faster and more controllable than gravimetric flow. This device allows for hands-off conversion, as enzymes will act on the blood as it travels through the column, limiting the human labor of the procedure. The columns are also disposable, which ensures sterility.
The airtight nature of the contraption was enforced with Luer lock connectors (see 5, Fig 19), a standard procedure that will allow us to securely connect the columns together. After the blood has been passed through all the columns, it will be collected in a collection centrifuge tube. A special narrow open cap (see 6, Fig 19) was designed to allow blood to flow directly from the columns into the collection tube while still being airtight. After processed blood has been collected into the tube, it can be detached from the columns, closed, and entered into the post-conversion processing step. Having several columns connected in series provides the added benefit of modularity. Depending on the blood type being converted, the columns with the appropriate enzymes can be assembled. There can thus be 3 different custom configurations of columns for type A, type B, and type AB blood.
In the second part of the conversion process, free antigens and any incompatible/uncleaved red blood cells are eliminated from the final product to ensure that the transfusion of processed blood does not result in adverse effects in the patients’ bloodstream. There is first a centrifugation and washing step, which allows for the separation of RBCs from impurities in the solution, such as residual antigens and enzymes from the previous conversion step. Following centrifugation, the supernatant containing the impurities will be decanted and then replaced with a clean saline solution. Then, antibody-antigen agglutination mechanisms separate cleaved and uncleaved RBCs. The appropriate antibody will be added to the washed solution. Uncleaved RBCs will react with the antibody and agglutinate, while cleaved RBCs will not. Because agglutinated clumps will be larger in size than individual RBCs, they can be filtered out using a leukodepletion filter. The final solution will thus only contain cleaved RBCs safe for transfusion.
Dimensions for the kit hardware were optimized to convert 1 unit of blood (taken to be 250 mL) at a time to avoid mixing blood from different donors (Fig 20). Column dimensions were determined to ensure the appropriate volume of enzyme-immobilized beads and blood volume would pass through the columns (1 unit of blood will be distributed throughout the 3 connected columns) based on rough estimates of enzyme efficiency. A 50 μm pore size was used for the leukocyte reduction filters to allow RBCs (7-8 μm) to pass through but prevent the 90 μm beads from flowing through (Kinnunen et al., 2011). Other dimensions were determined to ensure compatibility with industry norms.
Discussion
Long term trends of decreasing donor rates and increasing demand and short term developments like the COVID-19 pandemic have exposed the susceptibility of international and national blood organizations to shortages (Lemmens et al., 2005; Slonim et al., 2014; Stanworth et al., 2020). The purpose of this study was to explore the method of enzymatic blood type conversion to reduce ABO antigen transfusion incompatibility and thereby increase the supply of useful blood. Compared to alternative proposed solutions to increase blood supply, like commercialization and using artificial substitutes, such an approach has several distinct benefits. First, it avoids the ethical conflicts involved in uprooting the traditional voluntary system of blood donations. This approach also relies on converting existing RBCs rather than attempting to engineer synthetic RBCs from scratch, meaning it should be able to preserve native red blood cell function.
To evaluate our theory, we aimed to express, purify, and evaluate the functionality of 3 chosen recombinant enzymes and investigate how best to implement our project in a real blood center setting. As mentioned, these enzymes were chosen based on their ability to cleave their respective blood group antigens at neutral pH and at room temperature, properties that enable direct conversion of RBCs in blood and eliminate the need for incubation (Rahfeld & Withers, 2020; Higgins et al., 2009; Kwan et al., 2015; Liu et al., 2008; Liu et al., 2007). In the first part of our project, we were able to successfully purify the NAGA and α-gal enzymes. At first, we did not observe any clear visible bands in the SDS-PAGE protein gel. But the expected bands appeared after we made adjustments to our procedure, changing to an overnight IPTG induction and switching from lysing cell pellets with sonication to using lysis buffer.
However, despite our best efforts, we were unable to express or purify endo-β-gal. We derived the protein sequence from S. pneumoniae instead of C. perfringens, which optimizes enzyme activity (Kwan et al., 2015) and corrected errors in the protein sequence. In addition, we also added a 6x Histidine tag through a flexible Glycine-Serine linker upstream of the protein sequence, a T7 promoter and RBS (http://parts.igem.org/Part:BBa_K525998), and a double terminator (http://parts.igem.org/Part:BBa_B0015) to maximize our chances to express Endo-β-gal. Despite improvements to the sequences available on the iGEM database, we were unable to express it.
Speculations of factors that may have contributed to this include protein toxicity before induction, toxicity after induction, and the formation of inclusion bodies (Rosano, 2014). Initially, incompatibility with E. coli for endo-β-gal expression due to codon bias was considered. As such, the sequence was reoptimized for E. coli expression to eliminate this potential issue. However, issues with expression still arose during the experimental stage. To test for protein toxicity before induction, in which the protein is expressed prior to IPTG induction due to a leaky T7 construct and inhibits normal cell function, a control culture was grown along with a culture with added glucose. Theoretically, glucose would limit leaky expression; therefore, a relatively slow growth rate in the control culture would indicate protein toxicity before induction. No significant difference was detected between the glucose-treated culture and the control culture through OD600 measurements, which suggests that there was no toxicity before induction. A similar test was conducted to test for protein toxicity after induction, in which the protein is expressed after IPTG induction and inhibits normal cell function. A control culture was grown along with a culture induced with IPTG. A slower growth rate in the induced culture was expected if there was toxicity after induction. No significant difference was detected through OD600 measurements, which suggests that there was no protein toxicity after induction. Since no protein toxicity before or after induction was suggested by our tests, the last speculation for the failure of endo-β-gal expression is attributed to the formation of inclusion bodies, which may be caused by incorrect disulfide bond formation, incorrect folding in the protein, low solubility of the protein, or a missing post-translational modification which E. coli is incapable of performing. Although there was no measure to affirm or deny such a possibility, it remains our speculative reason for the failure to express endo-β-gal.
Proceeding with our experiments, we were able to successfully demonstrate the functionality and specificity of NAGA and α-gal through a variety of tests that included TLC, colorimetric tests, as well as mass spectrometry. We were able to verify the ability of the enzymes to cleave A-antigen and B-antigen trisaccharides in solution, demonstrating an initial proof of concept for enzymatic blood type conversion. We then moved on to testing on actual porcine RBCs.
We wanted to compare the degree of agglutination before and after the addition of enzymes as a marker for whether antigens were successfully cleaved off. We planned to use a hemagglutination assay for the experiment and score agglutination strength before and after on a standard 5-point scale for each well, which would then be converted to a modified marsh score (Moise Jr et al., 1995; Marsh, 1972). However, when we performed initial blood typing, we noticed that the agglutination strength when Anti-A was added, though clearly distinguishable, demonstrated only a 1+ agglutination score. When Anti-B was added, there was no visible agglutination. We hypothesize that this is because the anti-A and Anti-B serums obtained were too weak to produce strong agglutination, and the Anti-B serum did not have the specificity to recognize the α-gal antigen on porcine RBCs. We modified our protocol according to these observations.
Rather than observe RBCs using a hemagglutination assay, we used slide agglutination, which we found would display the difference in agglutination more visibly. We also increased enzyme reaction time and did not wash RBCs in PBS as it risked diluting the blood and altering consistency. With these changes, we were able to successfully show an observable decrease in agglutination following NAGA treatment of A type RBCs, suggesting blood type conversion did occur. To evaluate the ability of α-gal to cleave antigens on pRBCs, a different antibody agglutination mechanism with stronger and more specific antibodies may need to be employed.
Some limitations of this study include a need for more quantitative metrics that afford more sensitivity in measuring enzyme cleavage on RBCs beyond presence and absence. This will allow for the more accurate determination of the degree of enzyme cleavage when acting on actual antigens on the surface of RBCs and eventually allow for the determination of a turnover rate of blood type conversion in a given amount of time, from which the amount of enzyme needed for conversion can be optimized. Studies have suggested using hemagglutination assays, in which samples are treated to serially diluted antibody concentrations in U and V bottom wells, and the subsequent patterns formed on the wells can be observed and assigned a standardized score corresponding to agglutination strength (Moise Jr et al., 1995). This method is also advantageous as it sums the total scores of reaction in each well rather than just the minimum antibody “titer” needed for agglutination (Marsh, 1972). Another more quantitative method involves using microplate technology (Mujahid & Dickert, 2015). A study has proposed a passive cost-effective microfluidic device made of filter paper, whose results can then be captured through images and analyzed for agglutination using grayscale mean intensity through software like ImageJ, an image processing program (Samae et al., 2021).
Another limitation of this study is that it does not address the immunogenic effects from other antigen systems, most notably that of the Rh blood group system. The Rh system is determined by the presence or absence of the Rhesus antigen. Because the antigen is a transmembrane protein, we believe that the removal of the antigen through cleavage is implausible (Flegel, 2007). Indeed, other studies have instead opted for a “masking approach,” where polymers or single-stranded DNA aptamers are attached to block anti‐RhD recognition (Li et al., 2015; Zhang et al., 2020). After studying the literature on this subject, we decided not to pursue this topic and instead focus on ABO blood conversion, as we found it more achievable with the resources we were provided access to.
Lastly, we considered the feasibility of the implementation of our project. One limiting factor with this methodology is how efficient the enzymes used are. Hence, speaking with hematologists and blood bank specialists, we identified cost-effectiveness as a key consideration. With time and budget constraints, as well as concerns of wastage of RBCs, it is important that the cost, duration of conversion, and turnover efficiency of the prototype kit be optimized if a system like this is implemented effectively in the blood supply chain.
Next steps
In the next steps of this study, we hope to build on our current work by continuing to express and evaluate the function of our enzymes and begin performing experiments to validate the proposed implementation of our project. First, we hope to revisit the expression of endo-β-gal. To troubleshoot the expression and purification process, we plan to perform a closer comparison of the cataloged sequences across different databases, confirm the ability of E. coli to express the enzyme, alter the gene sequence to reduce hydrophobicity, or utilize fusion partners to increase solubility, assist folding with chaperones if necessary. Upon successful purification of endo-β-gal, we will subject the enzyme to a similar series of functional tests, such as performing TLC using lactose as a substrate (Jork et al., 1990). Next, we hope to obtain stronger and more specific antibodies, especially for human Anti-B, that will enable us to further evaluate the cleavage ability of both α-gal and NAGA on pRBCs using a hemagglutination assay as mentioned above, that will afford greater sensitivity in data collection.
We also hope to perform experiments that will potentially provide proof of concept for our blood type conversion prototype model. First, we aim to test enzyme cleavage when immobilized on the surface of nickel sepharose beads. To achieve this, we will perform the enzyme purification process but skip the elution step. Agglutination of pRBCs before and after addition to the columns will be evaluated to examine if cleavage did occur.
Testing cell viability is another experiment that can be performed. It is important to evaluate if the proposed procedure causes inadvertent RBC death. Hence, we propose using a dye exclusion assay with trypan blue dye, a stain that exclusively labels dead cells. Upon analysis with light microscopy, dead cells will be blue. The concentration of dead cells before and after enzyme treatment can be quantified with a hemocytometer, and percent viability can be calculated as the number of unstained cells divided by the total number of cells (Fang & Trewyn, 2012; Li et al., 2017).
Lastly, we hope to perform experiments to validate our proposed method for the purification of converted RBCs, which constitutes the second part of our proposed implementation. We plan on testing the ability of filter paper or wired mesh to separate agglutinated and non-agglutinated RBCs. One way to do this is to lay an antibody-treated filter paper on top of a testing paper and examine for bleed-through of agglutinated and non-agglutinated RBCs onto the second layer (Jarujamrus et al., 2012).
Author contributions
All authors performed literature background research. WHT and KH designed the DNA constructs. YK, SH, WHT, JL, AC, ES, EL, and LH performed protein expression and purification. WHT, AC, and JL designed the experimental protocols. WHT, YK, SH, EL, AC, JL, LH, AL, and SK performed enzyme function tests. SH, WHT, and AC conducted porcine blood agglutination. BC and AL implemented the blood supply and demand model. All authors engaged in data analysis. WHT and BC designed the conversion kit prototype. JL, WHT, BC, SH, and MT designed the original graphics and created the figures used in this work. All authors contributed to the production of the video. WHT, YK, SH, and BC drafted the manuscript. All authors approved the submitted version of the manuscript.
Acknowledgments
The authors would like to thank Mr. Jun-Hong Liu (劉俊宏) from the Taipei Blood Center, Dr. Jimmy Kang from the National Taiwan University Hospital blood bank, Dr. Gua-Che Lu (盧冠澈) from Keelung Hospital, Dr. Frank Hsiao from Far Eastern Memorial Hospital, Dr. George Chen (陳榮治), and Dr. Chuang Ching-Cheng (莊競程) from National Yang Ming Chiao Tung University (NYCU) for providing valuable insight that helped steer our project direction. We would also like to thank Dr. Todd Lowary and Louriel Macale from the Institute of Biological Chemistry at Academia Sinica for generously providing us with A and B antigen trisaccharides and access to and technical assistance with mass spectrometry. We would like to thank Dr. Michael Li (黎萬君) and Dr. Yi-ta Hsieh (謝宜達) from NYCU for providing us a space to conduct experiments when our school was closed because of COVID-19 and for allowing us to use their microplate reader. The help we received from James An of the TAS Robotics Department in developing our prototype CAD model is also greatly appreciated.
Lastly, we would like to thank the faculty and administration of Taipei American School and especially our four invaluable pillars: Mr. Sean Tsao for resource acquisition and supervision, Dr. Nicholas Ward for guidance on data analysis and modeling, and our primary PIs Dr. Jonathan Hsu and Mr. Jude Clapper for supporting us both scientifically and spiritually every step of the way, from the conception to the culmination of our project. We would like to wish the best for the retiring Mr. Clapper; his legacy lives on in all the students he has inspired throughout the years.
References
American Red Cross. (2022, January 11). Red Cross Declares First-ever Blood Crisis amid Omicron Surge. Red Cross. Retrieved April 20, 2022, from https://www.redcross.org/about-us/news-and-events/press-release/2022/blood-donors-needed-now-as-omicron-intensifies.html
Artificial Blood. (n.d.). Pacific Heart, Lung & Blood Institute. Retrieved April 20, 2022, from https://www.phlbi.org/divisions/blood-disorders/artificial-blood/
Blood Types Explained – A, B, AB and O | Red Cross Blood Services. (n.d.). Red Cross Blood Donation. Retrieved April 21, 2022, from https://www.redcrossblood.org/donate-blood/blood-types.html
Cooling, L. (2015). Blood Groups in Infection and Host Susceptibility. Clinical microbiology reviews, 28(3), 801-870. https://doi.org/10.1128/CMR.00109-14
Dean, L. (2005). Chapter 1: Blood and the cells it contains. In Blood Groups and Red Cell Antigens (Chapter 1). National Center for Biotechnology Information (US). https://www.ncbi.nlm.nih.gov/books/NBK2263/
Dean, L. (2005). Chapter 2: Blood group antigens are surface markers on the red blood cell membrane. In Blood Groups and Red Cell Antigens (Chapter 2). National Center for Biotechnology Information (US). https://www.ncbi.nlm.nih.gov/books/NBK2263/
Dean, L. (2005). Chapter 5: The ABO blood group. In Blood Groups and Red Cell Antigens (Chapter 5). National Center for Biotechnology Information (US). https://www.ncbi.nlm.nih.gov/books/NBK2267/
Fang, I. J., & Trewyn, B. G. (2012). Application of mesoporous silica nanoparticles in intracellular delivery of molecules and proteins. Methods in enzymology, 508, 41-59. https://doi.org/10.1016/B978-0-12-391860-4.00003-3
Flegel, W. A. (2007). The genetics of the Rhesus blood group system. Blood Transfusion, 5(2), 50-57. https://doi.org/10.2450/2007.0011-07
Held, P. (2007). Kinetic Analysis of β-Galactosidase Activity Using the PowerWave™ HT and Gen5™ Data Analysis Software. BioTek. https://www.biotek.com/resources/docs/B-Gal_Michaelis-Menten_App_Note.pdf
Higgins, M. A., Whitworth, G. E., Warry, N. E., Randriantsoa, M., Samain, E., Burke, R. D., Vocadlo, D. J., & Boraston, A. B. (2009). Differential Recognition and Hydrolysis of Host Carbohydrate Antigens by Streptococcus pneumoniae Family 98 Glycoside Hydrolases. THE JOURNAL OF BIOLOGICAL CHEMISTRY, 284(38), 26161–26173. https://doi.org/10.1074/jbc.M109.024067
Hsieh, H.-Y., Calcutt, M. J., Chapman, L. F., Mitra, M., & Smith, D. S. (2003). Purification and characterization of a recombinant α-N-acetylgalactosaminidase from clostridium perfringens. Protein Expression and Purification, 32(2), 309–316. https://doi.org/10.1016/j.pep.2003.08.007
Jarujamrus, P., Tian, J., Li, X., Siripinyanond, A., Shiowatana, J., & Shen, W. (2012). Mechanisms of red blood cells agglutination in antibody-treated paper. Analyst, 137(9), 2205-2210. https://pubs.rsc.org/en/content/articlelanding/2012/AN/c2an15798e
Jork, H., Funk, W., Fischer, W., & Wimmer, H. (1990). Thin-Layer Chromatography Reagents and Detection Methods (F. A. Hampson & J. A. Hampson, Trans.). Verlag Chemie. http://library.nuft.edu.ua/ebook/file/Jork1990.pdf
Kinnunen, M., Kauppila, A., Karmenyan, A., & Myllylä, R. (2011, Jul 1). Effect of the size and shape of a red blood cell on elastic light scattering properties at the single-cell level. Biomed Opt Express, 2(7), 1803-1814. 10.1364/BOE.2.001803
Kruve, A., Kaupmees, K., Liigand, J., Oss, M., & Leito, I. (2013, May 22). Sodium adduct formation efficiency in ESI source. Journal of Mass Spectrometry, 48(6), 695-702. https://doi.org/10.1002/jms.3218
Kwan, D. H., Constantinescu, I., Chapanian, R., Higgins, M. A., Kötzler, M. P., Samain, E., Boraston, A. B., Kizhakkedathu, J. N., & Withers, S. G. (2015). Toward Efficient Enzymes for the Generation of Universal Blood through Structure-Guided Directed Evolution. Journal of the American Chemical Society, 137(17), 5695-5705. https://doi.org/10.1021/ja5116088
Lee, H., Algaze, S., & Tan, E. (2020, August 10). Michaelis-Menten Kinetics. Chemistry LibreTexts. Retrieved April 26, 2022, from https://chem.libretexts.org/Bookshelves/Biological_Chemistry/Supplemental_Modules_(Biological_Chemistry)/Enzymes/Enzymatic_Kinetics/Michaelis-Menten_Kinetics
Lemmens, K. P. H., Abraham, C., Hoekstra, T., Ruiter, R. A. C., De Kort, W. L. A. M., Brug, J., & Schaalma, H. P. (2005, Jun). Why don’t young people volunteer to give blood? An investigation of the correlates of donation intentions among young nondonors. Transfusion, 45(6), 945-55. 10.1111/j.1537-2995.2005.04379.x
Li, C., Li, Z., Xun, S., Jiang, P., Yan, R., Chen, M., Hu, F., Rupp, R. A., Zhang, X., Pan, L., & Xu, J. (2017). Protection of the biconcave profile of human erythrocytes against osmotic damage by ultraviolet-A irradiation through membrane-cytoskeleton enhancement. Cell Death Discovery, 3, 17040. https://doi.org/10.1038/cddiscovery.2017.40
Li, L., Noumsi, G. T., Kwok, Y. Y. E., Moulds, J. M., & Scott, M. D. (2015). Inhibition of phagocytic recognition of anti-D opsonized Rh D1 RBC by polymer-mediated immunocamouflage. American Journal of Hematology, 90(12), 1165–1170. https://doi.org/10.1002/ajh.24211
Liu, Q. P., Sulzenbacher, G., Yuan, H., Bennett, E. P., Pietz, G., Saunders, K., Spence, J., Nudelman, E., Levery, S. B., White, T., Neveu, J. M., Lane, W. S., Bourne, Y., Olsson, M. L., Henrissat, B., & Clausen, H. (2007). Bacterial glycosidases for the production of universal red blood cells. Nature Biotechnology, 25(4), 454-464. https://doi.org/10.1038/nbt1298
Liu, Q. P., Yuan, H., Bennett, E. P., Levery, S. B., Nudelman, E., Spence, J., Pietz, G., Saunders, K., White, T., Olsson, M. L., Henrissat, B., Sulzenbacher, G., & Clausen, H. (2008). Identification of a GH110 subfamily of alpha 1,3-galactosidases: novel enzymes for removal of the alpha 3Gal xenotransplantation antigen. The Journal of Biological Chemistry, 283(13), 8545-54. https://doi.org/10.1074/jbc.M709020200
Long, C., Hara, H., Pawlikowski, Z., Koike, N., d’Arville, T., Yeh, P., Ezzelarab, M., Ayares, D., Yazer, M., & Cooper, D. K. C. (2009). Genetically engineered pig red blood cells for clinical transfusion: initial in vitro studies. Transfusion, 49(11), 2418–2429. https://doi.org/10.1111/j.1537-2995.2009.02306.x
Loua, A., Kasilo, O. M. J., Nikiema, J. B., Sougou, A. S., Kniazkov, S., & Annan, E. A. (2021, February 2). Impact of the COVID-19 pandemic on blood supply and demand in the WHO African Region. Vox Sanguinis, 116(7), 774-784. https://doi.org/10.1111/vox.13071
Marsh, W. L. (1972). Scoring of hemagglutination reactions. Transfusion, 12(5), 352-353. https://doi.org/10.1111/j.1537-2995.1972.tb04459.x
Meloncelli, P. J., & Lowary, T. L. (2010). Synthesis of ABO histo-blood group type I and II antigens. Carbohydrate research, 345(16), 2305–2322. https://doi.org/10.1016/j.carres.2010.08.012
Mitra, R., Mishra, N., & Rath, G. P. (2014). Blood groups systems. Indian Journal of Anaesthesia, 58(5), 524–528. https://pubmed.ncbi.nlm.nih.gov/25535412/
Mizuno, J. (2013, Aug). Use of microaggregate blood filters instead of leukocyte reduction filters to purify salvaged, autologous blood for re-transfusion during obstetric surgery. J Anesth, 27(4), 645-6. 10.1007/s00540-013-1579-7
Moise Jr, K. J., Perkins, J. T., Sosler, S. D., Brown, S. J., Saade, G., Carpenter Jr, R. J., Thorp, J. A., Ludomirski, A., Wilkins, I. A., Grannum, P. A., & Copel, J. (1995). The predictive value of maternal serum testing for detection of fetal anemia in red blood cell alloimmunization. American Journal of Obstetrics and Gynecology, 172(3), 1003-1009. https://doi.org/10.1016/0002-9378(95)90034-9
Moradi, S., Jahanian-Najafabadi, A., & Roudkenar, M. H. (2016). Artificial Blood Substitutes: First Steps on the Long Route to Clinical Utility. Clin Med Insights Blood Disord, (9), 33-41. 10.4137/CMBD.S38461
Mujahid, A., & Dickert, F. L. (2015). Blood Group Typing: From Classical Strategies to the Application of Synthetic Antibodies Generated by Molecular Imprinting †. Sensors, 16(1), 51. https://doi.org/10.3390/s16010051
New England Biolabs. (n.d.). Endoglycosidase Reaction Buffer Pack. https://international.neb.com/products/b0701-endoglycosidase-reaction-buffer-pack#Product%20Information
New England Biolabs. (n.d.). Typical Reaction Conditions for α-N-acetylgalactosaminidase (P0734). https://international.neb.com/protocols/2013/01/10/typical-reaction-conditions-p0734
NIH. (2022, March 24). Treatments for Blood Disorders – Treatments for Blood Disorders. National Heart, Lung, and Blood Institute. Retrieved April 20, 2022, from https://www.nhlbi.nih.gov/health/blood-bone-marrow-treatments
Rahfeld, P., & Withers, S. G. (2020, January 10). Toward universal donor blood: Enzymatic conversion of A and B to O type. J Biol Chem, 295(2), 325-34. 10.1074/jbc.REV119.008164
Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in Microbiology, 5. https://doi.org/10.3389/fmicb.2014.00172
Samae, M., Chatpun, S., & Chirasatitsin, S. (2021). Hemagglutination Detection with Paper–Plastic Hybrid Passive Microfluidic Chip. Micromachines, 12(12), 1533. https://doi.org/10.3390/mi12121533
Sarkar, S. (2008). Artificial blood. Indian Journal of Critical Care Medicine, 12(3), 140-44. 10.4103/0972-5229.43685
Slonim, R., Wang, C., & Garbarino, E. (2014, Spring). The Market for Blood. Journal of Economic Perspectives, 28(2), 117-96. American Economic Association. 10.1257/jep.28.2.177
Smith, D. M., Newhouse, M., Naziruddin, B., & Kresie, L. (2006, May). Blood groups and transfusions in pigs. Xenotransplantation, 13(3), 186-94. 10.1111/j.1399-3089.2006.00299.x
Smood, B., Hara, H., Schoel, L. J., & Cooper, D. K. (2019). Genetically-engineered pigs as sources for clinical red blood cell transfusion: What pathobiological barriers need to be overcome? Blood Reviews, 35, 7-17. 10.1016/j.blre.2019.01.003
Souid, A.-K., Rosales, L. G., & Aram, B. B. (2009, May 15). Utilizing Blood Bank Resources/Transfusion Reactions and Complications. Pediatric Emergency Medicine, 931-935. 10.1016/B978-141600087-7.50135-5
Stanworth, S. J., New, H. V., Apelseth, T. O., Brunskill, S., Cardigan, R., & Carolyn Doree. (2020, October 1). Effects of the COVID-19 pandemic on supply and use of blood for transfusion. The Lancet Hematology, 7(10), 756-64. https://doi.org/10.1016/S2352-3026(20)30186-1
Taiwan Blood Services Foundation. (2021, July 21). 2020 Annual Report. Annual Report – Taiwan Blood Services Foundation. Retrieved April 20, 2022, from https://www.blood.org.tw/Internet/english/List.aspx?uid=7681&pid=7679
Taiwan News. (2021, May 21). Taiwan blood supplies running at critically low levels. Taiwan News. Retrieved April 20, 2022, from https://www.taiwannews.com.tw/en/news/4207762
WHO. (2010). Voluntary blood donation: foundation of a safe and sufficient blood supply. NCBI. Retrieved April 20, 2022, from https://www.ncbi.nlm.nih.gov/books/NBK305666/
Zhang, Y., Xu, H., Wang, X., Wang, L., Liu, R., Li, L., & Zhou, H. (2020). Single‑strained DNA aptamers mask RhD antigenic epitopes on human RhD+ red blood cells to escape alloanti‑RhD immunological recognition. Molecular medicine reports, 21(4), 1841–1848. https://doi.org/10.3892/mmr.2020.10985